
IL SUNDAY TIMES: "IL VIA LIBERA DELLE AUTORITÀ BRITANNICHE AL VACCINO ARRIVERÀ ENTRO GIOVEDÌ, MENTRE LA DISTRIBUZIONE DOVREBBE…"
Da "Ansa"
 ASTRAZENECA
ASTRAZENECA
Il vaccino sviluppato da AstraZeneca e dall'Università di Oxford è "efficace al 95%", come quelli già approvati di Pfizer e Moderna, "ed è in grado di eliminare al 100%" i sintomi gravi che portano ai ricoveri per Covid-19. Lo dice al Sunday Times Pascal Soriot, ad di AstraZeneca.
Secondo il gionale, il via libera delle autorità britanniche al vaccino arriverà "entro giovedì" mentre la sua distribuzione dovrebbe avvenire a partire dal 4 gennaio. Fonte: qui
Covid, vaccino di AstraZeneca efficace al 95%: tempi e costi ridotti
Il vaccino di Oxford sviluppato dall'azienda farmaceutica AstraZeneca "è in grado di eliminare al 100% i sintomi gravi" e potrebbe essere usato già a gennaio
Il vaccino anti Covid sviluppato in collaborazione dall’azienda di biotecnologie AstraZeneca e dalla Università di Oxford è “efficace al 95%“. Lo ha dichiarato Pascal Soriot, amministratore delegato della società farmaceutica, sottolineando una percentuale di successo simile alla profilassi prodotta da Pfizer, con cui è iniziata il 27 dicembre la campagna vaccinale in Italia sugli operatori sanitari, e a quella prodotta da Moderna.
La formula brevettata a Oxford è stata sperimentata già sulla popolazione anziana, risultando efficace tra gli over 70, cioè quelle fasce più esposte alle complicanze dovute all’infezione da coronavirus.
Covid, vaccino di Oxford al via da gennaio in Uk
Il via libera all’inoculazione del prodotto da parte dell’agenzia del farmaco britannica dovrebbe arriverà “entro giovedì” 31 dicembre, mentre la distribuzione nel Regno Unito dovrebbe iniziare già a partire dal 4 gennaio.
Vaccino di Oxford e AstraZeneca: le sue caratteristiche
Secondo quanto ha riferito il manager, il vaccino di Oxford, sviluppato in collaborazione con il nostro Paese, “è in grado di eliminare al 100%” i sintomi gravi di Covid-19, che spesso portano i pazienti a essere ricoverati in terapia intensiva e in rianimazione.
Il vaccino prodotto da AstraZeneca e dalla Università di Oxford è una “formula vincente“, ha sottolineato l’amministratore delegato Pascal Soriot. “Finora riteniamo che il vaccino sia efficace anche contro la variante inglese del virus, ma non possiamo esserne certi, così condurremo dei test”.
Covid: perché il vaccino di AstraZeneca costa di meno
Rispetto al vaccino prodotto da Pfizer e BioNTech, quello di AstraZeneca ha un punto di forza per quanto riguarda la logistica: può essere immagazzinato, trasportato e maneggiato a temperature superiore. Richiede infatti condizioni di refrigerazione tra i 2 e gli 8 gradi centigradi, ben al di sopra dei -70° C richiesti dal concorrente.
Grazie a questa particolarità, i costi potrebbero essere abbattuti, considerando che la catena del freddo potrebbe essere rispettata grazie ad apparecchiature già ampiamente usate per altre tipologie di vaccino.
Inoltre AstraZeneca si è impegnata a distribuirla al costo di produzione, ovvero 2,8 euro a dose. Si tratta di circa un decimo del prezzo di mercato di Moderna e Pfizer. Per questo potrà essere facilmente acquistato e utilizzato nei Paesi più poveri. Fonte: qui
Il vaccino AZD1222 per il COVID-19 ha prodotto importanti risposte immunitarie su tutti i partecipanti dello studio clinico di fase I/II
I dati intermedi hanno mostrato forti risposte degli anticorpi e dei linfociti T
Milano, 20 luglio 2020 – I risultati intermedi dello studio clinico di fase I/II COV001 – tuttora in corso e condotto dall’Università di Oxford – hanno dimostrato che AZD1222 è stato ben tollerato e ha generato importanti risposte immunitarie contro il virus SARS-CoV-2 in tutti i partecipanti arruolati.
COV001 è uno studio clinico di fase I/II, multicentrico, randomizzato, controllato e condotto in cieco su 1.077 partecipanti adulti sani di età compresa tra i 18 e i 55 anni. Lo studio ha valutato una singola dose di AZD1222 confrontandolo con MenACWY, un vaccino meningococcico coniugato. Dieci partecipanti hanno anche ricevuto due dosi di AZD1222 a distanza di un mese.
I risultati pubblicati su The Lancet hanno confermato che una singola dose di AZD1222 ha prodotto, un mese dopo l’iniezione, un aumento di quattro volte degli anticorpi contro la proteina spike del virus SARS-CoV-2, nel 95% dei partecipanti. In tutti i partecipanti è stata indotta una risposta via linfociti T, che ha raggiunto il picco entro il quattordicesimo giorno e si è mantenuto fino a due mesi dopo l’iniezione.
L’attività neutralizzante contro il SARS-CoV-2 (come valutato dal saggio MNA80) è stata osservata nel 91% dei partecipanti un mese dopo la vaccinazione, e nel 100% dei partecipanti che avevano ricevuto una seconda dose. I livelli di anticorpi neutralizzanti, osservati nei partecipanti che avevano ricevuto una o due dosi, erano compresi in un intervallo simile a quelli osservati nei pazienti COVID-19 convalescenti. Sono state osservate forti correlazioni tra i saggi di neutralizzazione.
Le prime risposte di sicurezza hanno confermato che le reazioni locali e sistemiche transitorie erano comuni nel gruppo AZD1222 ed erano paragonabili a quelle osservate in studi precedenti e in altri vaccini vettoriali adenovirali1,2,3,4. Esse includevano temporaneo dolore e sensibilità al sito di iniezione, mal di testa da lieve a moderato, affaticamento, brividi, febbre, malessere e dolori muscolari. Non sono stati segnalati eventi avversi gravi con AZD1222 e le reazioni sono state ridotte dall'uso del paracetamolo profilattico, e si sono verificate meno frequentemente dopo una seconda dose.
Il professor Andrew Pollard, Chief investigator dell’Oxford Vaccine Trial presso la Oxford University e coautore dello studio, ha dichiarato: “I dati intermedi di fase I/II dimostrano che il nostro vaccino contro il coronavirus non ha prodotto reazioni impreviste e ha avuto un profilo di sicurezza simile a quello di precedenti vaccini di questo tipo. Le risposte immunitarie osservate dopo la vaccinazione sono in linea con ciò che prevediamo sarà associato alla protezione contro il virus SARS-CoV-2, anche se per confermarlo dobbiamo continuare il nostro rigoroso programma di studi clinici. La risposta immunitaria più importante è stata osservata nei partecipanti che hanno ricevuto due dosi di vaccino, il che indica che questa potrebbe essere una buona strategia vaccinale”.
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, ha affermato: “I dati intermedi di fase I/II – che dimostrano come AZD1222 sia stato in grado di generare una rapida risposta via anticorpi e linfociti T contro il virus SARS-CoV-2 – sono per noi molto incoraggianti. Sebbene vi sia ancora molto lavoro da fare, i dati odierni ci consentono sia di confidare maggiormente nell’efficacia del vaccino, sia di continuare i nostri piani per la produzione su larga scala, con l’obiettivo di favorire un accesso ampio ed equo in tutto il mondo”.
Attualmente studi avanzati di fase II/III sono in corso nel Regno Unito, in Brasile e in Sudafrica, e stanno per iniziare negli Stati Uniti. Queste sperimentazioni determineranno l’entità della protezione dal COVID-19 che il vaccino sarà in grado di offrire, e misureranno la sicurezza e le risposte immunitarie nelle diverse fasce di età e a seconda dei vari dosaggi.
Parallelamente, AstraZeneca continua ad adempiere al proprio impegno per un ampio ed equo accesso al vaccino, nel caso in cui gli studi clinici in fase avanzata risultino efficaci. Finora è stata concordata la fornitura di oltre due miliardi di dosi di vaccino a Regno Unito, Stati Uniti, Inclusive Vaccines Alliance in Europa, Coalition for Epidemic Preparedness, Gavi the Vaccine Alliance e Serum Institute of India.
Note per i redattori
Considerazioni economiche
L’odierna comunicazione non dovrebbe avere alcun impatto sull’orientamento finanziario dell’azienda per il 2020, poiché si prevede che le spese per lo sviluppo del vaccino saranno compensate dai finanziamenti di governi e dalle organizzazioni internazionali.
I correlati immunitari di protezione contro il COVID-195
I correlati di protezione per un vaccino contro il COVID-19 non sono stati ancora definiti. Nei soggetti che si sono ripresi dall’infezione da SARS-CoV-2 è stata dimostrata la presenza di livelli elevati di anticorpi neutralizzanti. Inoltre, i dati emergenti suggeriscono che una risposta via linfociti T potrebbe svolgere un ruolo importante nella mitigazione della malattia. Alcuni individui che sono stati infettati dal virus, ma sono rimasti asintomatici, hanno sviluppato una robusta risposta dei linfociti T, con assenza di anticorpi rilevabili. La rapida induzione di anticorpi e linfociti T contro il virus SARS-CoV-2 potrebbe essere importante ai fini della protezione contro il COVID-19.
COV001
COV001 è uno studio di fase I/II controllato, randomizzato, condotto in cieco su 1.077 adulti sani in cinque centri di sperimentazione del Regno Unito al fine di determinare la sicurezza, l’immunogenicità e l’efficacia di AZD1222, candidato vaccino contro il COVID-19. I partecipanti – di età compresa tra i 18 e i 55 anni – hanno ricevuto una o due dosi di AZD1222 a 5x1010 particelle virali, oppure una singola dose di un vaccino meningococcico coniugato MenACWY, come vaccino di confronto.
Ai partecipanti sono stati prelevati campioni di sangue e sono state effettuate valutazioni cliniche per sicurezza e immunogenicità ai giorni 0 e 28, cui faranno seguito altri prelievi e valutazioni ai giorni 184 e 364. Inoltre, i partecipanti arruolati nella componente di fase I dello studio e nei due gruppi di dosaggio sono stati sottoposti a visite mediche ai giorni 3, 7, 14 e 28 dopo ogni vaccinazione.
AZD1222
AZD1222 è stato sviluppato congiuntamente dall’Università di Oxford e dalla sua società affiliata, Vaccitech. Utilizza un vettore virale di scimpanzé con deficit di replicazione basato su una versione indebolita di un comune virus del raffreddore (adenovirus) che causa infezioni negli scimpanzé e contiene il materiale genetico della proteina spike SARS-CoV-2. Dopo la vaccinazione, viene prodotta la proteina spike superficiale, la quale attiva il sistema immunitario affinché attacchi il SARS-CoV-2, se questo dovesse in seguito infettare l’organismo.
AstraZeneca
AstraZeneca è un’azienda biofarmaceutica globale orientata all’innovazione e focalizzata su scala internazionale nella ricerca scientifica, nello sviluppo e nella commercializzazione di farmaci con obbligo di prescrizione medica per patologie cardiovascolari, metaboliche, respiratorie, infiammatorie, autoimmuni, oncologiche, infezioni e disturbi del sistema nervoso centrale. AstraZeneca opera in oltre 100 Paesi e i suoi farmaci innovativi sono utilizzati da milioni di pazienti nel mondo. Maggiori informazioni su: http://www.astrazeneca.it
***
Per maggiori informazioni:
Ufficio stampa – Noesis
Samanta Iannoni – M: +39 348 1511488 - samanta.iannoni@noesis.net
Valeria Riccobono – T: +39 02 83105195 - valeria.riccobono@noesis.net
Lodovica Villa – M: +39 338 9131137 - lodovica.villa@noesis.net
References
1. Antrobus, R.D., et al., Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved Influenza A antigens. Mol Ther, 2014. 22(3): p. 668-74.
2. Coughlan, L., et al., Heterologous Two-Dose Vaccination with Simian Adenovirus and Poxvirus Vectors Elicits Long-Lasting Cellular Immunity to Influenza Virus A in Healthy Adults. EBioMedicine, 2018.
3. Wilkie, M., et al., A phase I trial evaluating the safety and immunogenicity of a candidate tuberculosis vaccination regimen, ChAdOx1 85A prime - MVA85A boost in healthy UK adults. Vaccine, 2020. 38(4): p. 779-789.
4. Folegatti, P.M., et al., Safety and immunogenicity of a candidate Middle East respiratory syndrome coronavirus viral-vectored vaccine: a dose-escalation, open-label, non-randomised, uncontrolled, phase 1 trial. The Lancet Infectious Diseases, 2020.
5. Sekine, T., et al., Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. BioRxiv, 2020. preprint doi: https://doi.org/10.1101/2020.06.29.174888.
Nuovo coronavirus: il punto sui vaccini in sperimentazione
Una parte importante della comunità scientifica sta lavorando sulla predisposizione di un vaccino efficace contro Covid-19. Questo articolo riporta gli ultimi aggiornamenti in materia.
Indice degli argomenti
Ultime novità
- mRNA-1273: l'FDA rilascia l'autorizzazione all'uso di emergenza per il vaccino Moderna
- ChAdOx1 nCoV-19: su The Lancet pubblicato il protocollo del vaccino
- AZD1222: AstraZeneca rende noti i dati di efficacia del suo vaccino
- BNT162b2: l'FDA rilascia l'autorizzazione all'uso di emergenza per il vaccino Pfizer-BioNTech, l'EMA lo raccomanda per l'autorizzazione anche in UE
- Gam-COVID-Vac: resi noti i dati di efficacia del vaccino Sputnik V
Su quali tipologie di vaccini si sta lavorando
La
Coalition for Epidemic Preparedness and Innovations (CEPI), organizzazione internazionale che ha lo scopo di promuovere lo
sviluppo e lo
stoccaggio di
vaccini contro microorganismi in grado di causare nuove e spaventose epidemie, sta
coordinando i numerosi progetti per la preparazione di vaccini contro il virus SARS-CoV-2.
A causa della recente scoperta del virus e della difficoltà di prevedere il tipo di risposta immunitaria prodotta, le strategie adottate risultano molto diversificate fra loro e, di conseguenza, il tipo di vaccino in grado di proteggere dall’infezione.
In particolare, i ricercatori stanno lavorando su tre tipologie di vaccini:
- Vaccino a RNA: si tratta di una sequenza di RNA sintetizzata in laboratorio che, una volta iniettata nell’organismo umano, induce le cellule a produrre una proteina simile a quella a quella verso cui si vuole indurre la risposta immunitaria (producendo anticorpi che, conseguentemente, saranno attivi contro il virus)
- Vaccino a DNA: il meccanismo è simile al vaccino a RNA. In questo caso viene introdotto un frammento di DNA sintetizzato in laboratorio in grado d’indurre le cellule a sintetizzare una proteina simile a quella verso cui si vuole indurre la risposta immunitaria
- Vaccino proteico: utilizzando la sequenza RNA del virus (in laboratorio), si sintetizzano proteine o frammenti di proteine del capside virale. Conseguentemente, iniettandole nell’organismo combinate con sostanze che esaltano la risposta immunitaria, si induce la risposta anticorpale da parte dell’individuo.
Come funziona la sperimentazione clinica di un vaccino
Nonostante la forte pressione esercitata dalla pandemia di COVID-19, e la speranza che ognuno di noi ripone nella ricerca scientifica, il futuro utilizzo di un vaccino deve essere necessariamente preceduto da studi rigorosi che richiedono il tempo necessario per valutarne l’efficacia e la sicurezza.
Inizialmente la ricerca ha inizio con la valutazione in vitro delle componenti dell’agente che andrà a costituire la componente attiva del vaccino.
Una volta definito questo aspetto ha inizio la cosidetta fase preclinica in cui viene testata la risposta immunitaria e/o i meccanismi avversi su organismi viventi complessi non umani.
Superata questa fase ha inizio la vera e propria sperimentazione clinica sull'uomo, che normalmente inizia dopo circa 2-5 anni dalle iniziali ricerche sulla risposta immunitaria, cui seguono altri 2 anni di prove pre-cliniche che coinvolgono la sperimentazione animale. La sperimentazione clinica si realizza in 3 fasi, in base al modello sperimentale adottato, la quantità di componente somministrata e la numerosità del campione di popolazione coinvolta:
- Fase I: prima somministrazione del vaccino sull’uomo per valutare la tollerabilità e la sicurezza del prodotto (il numero dei soggetti coinvolti è molto ridotto)
- Fase II: se la fase I ha mostrato risultati positivi, il vaccino viene somministrato ad un numero maggiore di soggetti (sempre eseguo) per valutare la risposta immunitaria prodotta, la tollerabilità, la sicurezza e definire le dosi e i protocolli di somministrazione più adeguati.
- Fase III: se la fase II ha mostrato risultati soddisfacenti, il vaccino viene somministrato a un numero elevato di persone allo scopo di valutare la reale funzione preventiva del vaccino.
Se tutte le fasi hanno dato esito positivo, il vaccino viene registrato e si procede alla produzione e distribuzione su larga scala.
Lo sviluppo del vaccino è un processo lungo, che normalmente richiede anni e numerosi investimenti economici. I trial clinici richiedono molti test su migliaia di persone e normalmente iniziano dopo circa 2-5 anni dalle iniziali ricerche sulla risposta immunitaria, cui seguono altri due anni di prove precliniche che coinvolgono la sperimentazione animale.
Se il vaccino risulta sicuro ed efficace, deve poi rispondere a tutti i requisiti regolatori e ottenere l'approvazione. Nell'attuale emergenza, è stato proposto un periodo di tempo più ristretto compreso tra 12 e 18 mesi, con team di esperti di tutto il mondo che lavorano per aumentare la velocità per trovare un candidato efficace.
Inoltre, trattandosi di un’emergenza sanitaria che interessa tutto il mondo, la capacità di produzione dovrebbe essere garantita prima del termine degli studi clinici e ripartita globalmente per garantirne anche un'equa distribuzione. A tal proposito, l'OMS ha riunito leader mondiali e partner sanitari, compresi quelli del settore privato, in un'iniziativa mirata ad accelerare lo sviluppo e la produzione del nuovo vaccino anti Covid-19, di test e trattamenti per consentire un accesso equo in tutto il mondo.
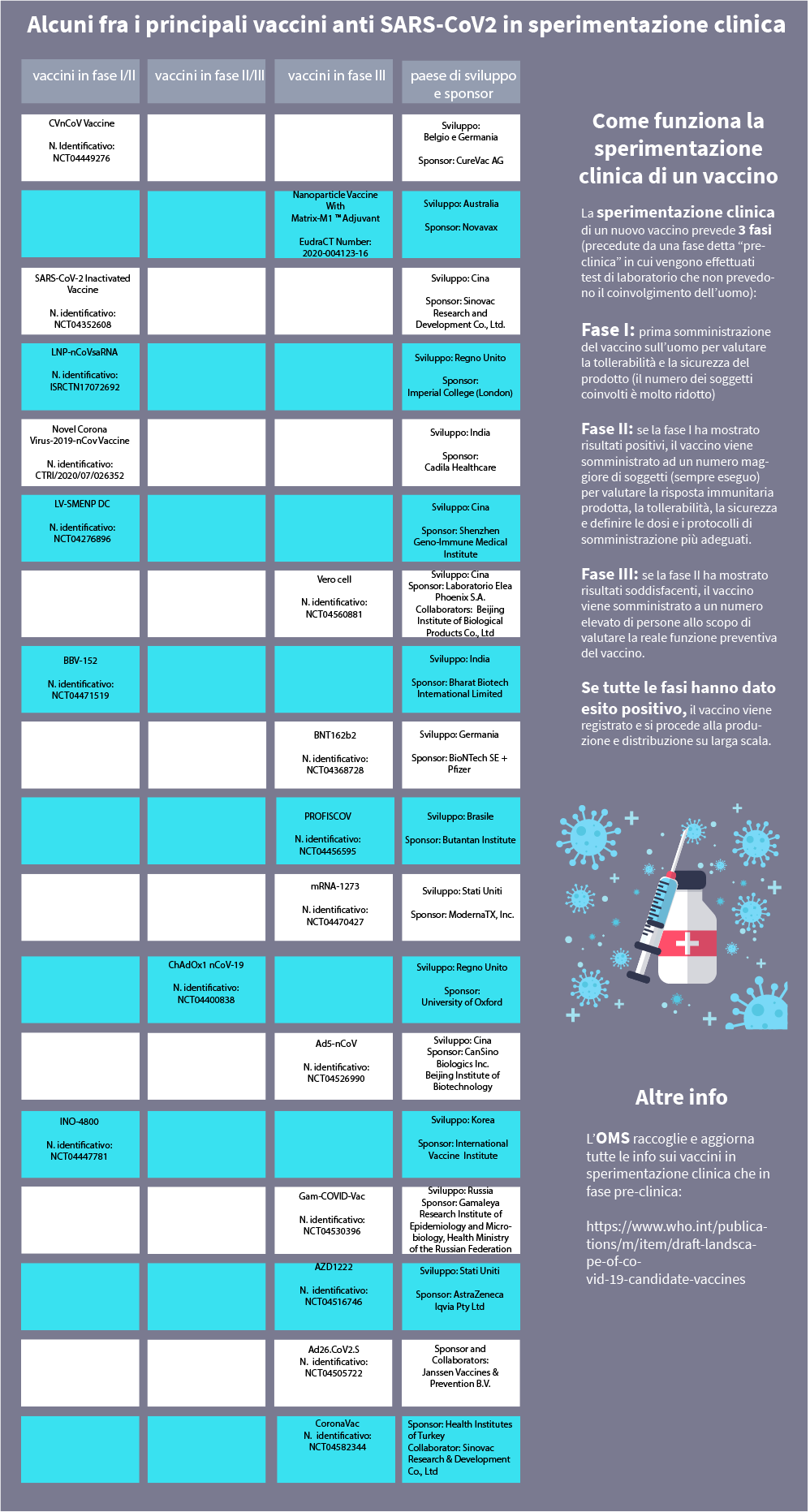
| Fra i numerosi studi attivati presso i più importanti istituti di ricerca di tutto il mondo, segnaliamo prendendo a riferimento il sito www.clinicaltrials.gov, trial clinici finalizzati a valutare l’efficacia di vaccini contro l’infezione da SARS-CoV2 (COVID-19). Per rimanere costantemente aggiornati è possibile controllare sulla pagina Coronavirus disease (COVID-2019) R&D dell'Organizzazione mondiale della sanità i report sullo stato delle ricerche. |
A che punto siamo con il vaccino contro il coronavirus
La ricerca è in continua evoluzione: riportiamo in forma grafica e nel testo sotto i trial più significativi, per ciascuna fase di sperimentazione in atto.
BBV152 (numero identificativo: NCT04471519)
Lo studio Whole-Virion Inactivated SARS-CoV-2 Vaccine (BBV152) for COVID-19 in Healthy Volunteers (BBV152) ha l’obiettivo di valutare la sicurezza, la reattogenicità, la tollerabilità e l'immunogenicità di tre gruppi di vaccini sperimentali e un gruppo placebo in volontari sani che ricevono due dosi intramuscolari di formulazioni di vaccino BBV152. L'obiettivo finale è la selezione di due formulazioni di vaccini che siano sicuri, ben tollerati e immunogenici, che saranno ulteriormente valutati nello studio di fase 2. E’ previsto l’arruolamento di 375 soggetti nella fase 1 e di 750 nella fase 2.
Sponsor: Bharat Biotech International Limited
Collaborator: Indian Council of Medical Research
CVnCoV Vaccine (numero identificativo: NCT04449276)
Sponsor: CureVac AG
Collaborator: Coalition for Epidemic Preparedness Innovations (CEPI)
INO-4800 (numero identificativo: NCT04447781)
Lo studio di fase 1/2
Safety, Tolerability and Immunogenicity of INO-4800 for COVID-19 in Healthy Volunteers ha come obiettivo quello di valutare la sicurezza, la tollerabilità e il profilo immunologico di INO-4800 somministrato mediante iniezione intradermica (ID) seguita da elettroporazione (EP) utilizzando il dispositivo CELLECTRA® 2000 in adulti sani di età compresa tra 19 e 64 anni in Corea. E’ previsto l’arruolamento di 160 soggetti che riceveranno INO-4800 1mg / dose + EP utilizzando CELLECTRA® 2000 (dosaggio al giorno 0 e alla 4
a settimana).
Sponsor: International Vaccine Institute
Collaborators: Coalition for Epidemic Preparedness Innovations, Inovio Pharmaceuticals
LNP-nCoVsaRNA (numero identifcativo: ISRCTN17072692)
Quando viene iniettato nel muscolo, il vaccino innesca le cellule a produrre copie della proteina spike e queste stimolano il corpo a produrre anticorpi.
Sono coinvolti adulti sani di età compresa tra 18 e 45 anni (per l'incremento della dose e la valutazione) e di età compresa tra 18 e 75 anni (per la valutazione della sicurezza).
Per l'aumento della dose, i partecipanti ricevono 0,1 µg di vaccino e sono invitati a registrare le informazioni su eventuali reazioni locali e sistemiche in un diario online la sera e successivamente ogni giorno per 6 giorni. Dopo 48 ore il team chiamerà il primo partecipante e controllerà il diario. Se non ci sono problemi di sicurezza dopo 48 ore, la dose del vaccino verrà gradualmente incrementata in pazienti successivi fino alla dose più alta (1,0 µg). I partecipanti saranno seguiti per 52 settimane in totale. Per la valutazione randomizzata della dose, i partecipanti vengono assegnati in modo casuale alle tre diverse dosi e vengono seguiti per un totale di 52 settimane. Per la valutazione della sicurezza estesa non randomizzata, i partecipanti ricevono la dose più alta (1,0 µg).
Sponsor information: Imperial College London
LV-SMENP-DC (numero identificativo: NCT04276896)
Il vaccino LV-SMENP-DC viene preparato ingegnerizzando le cellule dendritiche con il vettore lentivirale che esprime i domini conservati delle proteine strutturali SARS-CoV-2 e la proteasi utilizzando i minigeni SMENP. I linfociti T citotossici saranno attivati da LV-DC che presentano antigeni specifici per Covid-19. Il soggetto riceve rispettivamente un totale di 5x10 ^ 6 cellule di vaccino LV-DC e 1x10 ^ 8 linfociti T citotossici specifici per l'antigene tramite iniezione sottocutanea e infusione endovenosa. I pazienti vengono seguiti settimanalmente per un mese dopo l'infusione, mensilmente per 3 mesi e poi ogni 3 mesi fino al termine dello studio.
Sponsor: Shenzhen Geno-Immune Medical Institute
Collaborators: Shenzhen Third People's Hospital, Shenzhen Second People's Hospital
Novel Corona Virus-2019-nCov Vaccine (numero identificativo: CTRI/2020/07/026352)
E’ previsto l’arruolamento di soggetti sani di sesso maschile e femminile non in gravidanza, non in allattamento di età compresa tra 18 e 55 anni di peso corporeo> 50 kg per i maschi e> 45 kg per le femmine e BMI compreso tra 18,5 e 29,9 kg / m2. I soggetti maschi e femmine in età fertile devono praticare una contraccezione altamente efficace durante lo studio ed essere disposti e in grado di continuare la contraccezione per 90 giorni dopo somministrazione dell'ultimo vaccino in studio. E’ prevista la somministrazione di vaccino nCov alla dose di -0,1 ml 1 sola volta al giorno 0 e al giorno 28 e giorno 56.
Sponsor: Cadila Healthcare
SARS-CoV-2 Inactivated Vaccine (numero identificativo: NCT04352608)
Lo studio di fase I
Safety and Immunogenicity Study of Inactivated Vaccine for Prophylaxis of SARS CoV-2 Infection (COVID-19 è randomizzato, in doppio cieco, monocentrico, controllato con placebo in adulti di età compresa tra 18 e 59 anni. Lo scopo di questo studio è valutare l'immunogenicità e la sicurezza del vaccino sperimentale SARS-CoV-2 inattivato. E’ previsto l’arruolamento di 744 soggetti, di cui 144 nella fase I e 600 nella fase II. Il partecipante verrà assegnato a ricevere due dosi di vaccino sperimentale o placebo secondo la schedula vaccinale 0,14 o 0,28.
Sponsor: Sinovac Research and Development Co., Ltd.
Information provided by (Responsible Party): Sinovac Biotech Co., Ltd ( Sinovac Research and Development Co., Ltd. )
ChAdOx1 nCoV-19 (codice identificativo: NCT04400838)
NOVITA' Pubblicato su The Lancet il protocollo del vaccino
In sintesi lo studio comprende 4 coorti:
1) COV001 (Regno Unito): sperimentazione clinica di fase 1/2 in singolo cieco effettuata in cinque centri di ricerca del Regno Unito, iniziata il 23 aprile 2020 che ha previsto l’arruolamento 1.077 volontari sani di età compresa tra 18 e 55 anni. I partecipanti sono stati assegnati in modo casuale con rapporto 1:1 a ricevere ChAdOx1 nCoV-19 a una dose di 5 × 10¹⁰ particelle virali (dose standard), misurata utilizzando spettrofotometria, o vaccino di controllo (MenACWY : meningococcico gruppo A, C, W e Y). A un sottogruppo in aperto non randomizzato di dieci partecipanti, sono state somministrate due dosi di ChAdOx1 nCoV-19 a 28 giorni di distanza, come riportato in precedenza. Questa coorte era stata originariamente pianificata a dose singola e 88 partecipanti nella parte di fase 1 del studio rimangono destinatari di una singola dose. Tuttavia, a seguito delle robuste risposte immunitarie osservate nelle coorti con richiamo, il protocollo è stato modificato in un regime a due dosi.
2) COV002 (Regno Unito): sperimentazione clinica di fase 2/3 in singolo cieco, iniziata il 28 maggio 2020, che ha previsto l’arruolamento di 10.673 ad elevato rischio di esposizione a SARS-CoV-2 (operatori sanitari o sociali) provenienti da 19 centri localizzati in Inghilterra, Galles e Scozia.
In questa coorte sono presenti due gruppi di dosaggio: partecipanti che hanno ricevuto una primo vaccino a bassa dose (Low Dose, LS) (2•2 × 10¹⁰ particelle virali) seguito da una seconda dose standard (SD) (5 × 10¹⁰ particelle virali) definito come gruppo LD/SD; partecipanti che hanno ricevuto due dosi standard di vaccino (5 × 10¹⁰ particelle virali) sia nella prima che nella seconda somministrazione (gruppo SD/SD). Come spiegano gli Autori, tale differenza è dovuta al fatto che durante i controlli di qualità dei lotti di produzione, sono state osservate delle differenze nel dosaggio legate alle diverse metodiche di quantificazione adottate dai centri di produzione (spettrofotometria e PCR quantitativa [qPCR]). A seguito di questa rilevazione, in accordo con l’Ente nazionale di controllo (Medicines and Healthcare products Regulatory Agency), è stata selezionata la dose di 5 × 10¹⁰ particelle virali mediante spettrofotometro (2•2 × 10¹⁰ particelle virali da qPCR), per essere coerente con lo studio di fase 1 (COV001) condotto con l'uso della spettrofotometria.
Dopo la revisione e l'approvazione da parte del regolatore, il gruppo di studio ha concluso che la rilevazione mediante qPCR (a basso dosaggio) era più accurata e le dosi successive sono state adeguate alla dose standard (5 × 10¹⁰ particelle virali) utilizzando la metodica qPCR. Il protocollo è stato modificato il 5 giugno 2020, con conseguente arruolamento di due distinti gruppi con diversi regimi di dosaggio senza pause nell’arruolamento. Attualmente è stata sviluppata una procedura di analisi delle dosi e tutti i lotti vengono rilasciati solamente con una dose specifica di 3•5–6•5 × 10¹⁰ particelle virali (utilizzato per le dosi di richiamo nell'analisi di efficacia presentata nell’articolo).
La coorte LD/SD (di età compresa tra 18 e 55 anni) è stata arruolata per 11 giorni tra il 31 maggio e il 10 giugno 2020. La coorte SD/SD (di età compresa tra 18 e 55 anni) è stata arruolata dal 9 giugno al 20 luglio 2020. Successivamente, sono state arruolate coorti di età ≥ di 56 anni.
3) COV003 (Brasile): sperimentazione di fase 3 in singolo cieco iniziato il 23 giugno 2020. I partecipanti erano soggetti di età superiore ai 18 anni (N=10.002) ad alto rischio d’infezione da SARS-CoV-2 (operatori sanitari) provenienti da 6 centri brasiliani. A tutti i partecipanti sono state offerte due dosi di vaccino di 3•5–6•5 × 10¹⁰ particelle virali con somministrazione fino a 12 settimane di distanza (target 4 settimane).
4) COV005 (Sud Africa): sperimentazione in fase 1/2 in doppio cieco in corso in Sud Africa su adulti sani di età compresa tra 18 e 65 anni arruolati a partire dal 28 giugno 2020 (N=2.096). A tutti i partecipanti sono state offerte due dosi del vaccino di 3•5–6.5 × 10¹⁰ particelle virali somministrate a 4 settimane di distanza.
I tempi di somministrazione del vaccino (prima dose e richiamo) variano tra gli studi. A causa degli emendamenti descritti in precedenza, non è stato possibile uniformare a 4 settimane la somministrazione della seconda dose. Nella coorte COV002, 1.459 (53,2%) di 2.741 partecipanti del gruppo LD/SD hanno ricevuto una seconda dose dopo almeno 12 settimane dalla prima e solo 22 (0,8%) hanno ricevuto una seconda dose entro 8 settimane dalla prima. L'intervallo mediano tra le dosi per il gruppo SD/SD in COV002 è stato di 69 giorni. Al contrario, la maggioranza dei partecipanti in COV003 ha ricevuto una seconda dose entro 6 settimane dalla prima.
In tutti e quattro gli studi, il vaccino ha avuto una profilo di sicurezza buono, con eventi avversi gravi ed eventi avversi di interesse speciale bilanciati tra i bracci dello studio. Gravi eventi avversi si sono verificati in 168 partecipanti, 79 dei quali avevano ricevuto ChAdOx1 nCoV-19 e 89 dei quali avevano ricevuti MenACWY o controllo salino. I principali eventi avversi segnalati sono stati: un caso di mielite trasversa, un caso di febbre superiore a 40° C. Tutti i partecipanti alla sperimentazione si sono ripresi o si trovano in condizioni stabili o di miglioramento. Sono stati segnalati quattro decessi non COVID-19 negli studi (tre nel braccio di controllo e uno nel ChAdOx1 nCoV-19) per cause violente (incidente stradale, trauma contusivo, omicidio e polmonite fungina).
Rispetto all’outcome principale, riguardante la valutazione di efficacia del vaccino ChAdOx1 nCoV-19, i risultati mostrano un’efficacia significativa del 70,4% dopo due dosi e una protezione del 64,1% dopo almeno uno dose standard, contro la malattia sintomatica, senza problemi di sicurezza. Inizialmente, il gruppo di lavoro aveva sollevato la preoccupazione che il regime LD/SD potesse avere un'efficacia inferiore rispetto a SD/SD e l'accettazione da parte delle autorità regolatorie dell'inclusione dei due regimi di vaccinazione (LD/SD e SD/SD) nell'analisi si è basata sull’osservazione che questi regimi hanno generato livelli simili di anticorpi aumentando, così, la dimensione del campione disponibile per l'analisi senza comprometterne l'efficacia.
Nei partecipanti che hanno ricevuto due dosi standard, l'efficacia contro il COVID-19 sintomatico primario è stata coerente sia nel Regno Unito (efficacia del 60,3%) che in Brasile (efficacia del 64,2%), indicando che questi risultati sono generalizzabili in due diverse impostazioni con tempi diversi per la dose di richiamo (con la maggior parte dei partecipanti nel Regno Unito che riceve la dose di richiamo più di 12 settimane dopo la prima dose e la maggior parte dei partecipanti in Brasile che riceve la seconda dose entro 6 settimane dalla prima). L'efficacia del 90,0% osservata in coloro che hanno ricevuto una prima dose a basso dosaggio (Regno Unito) è stata sorprendentemente alta rispetto agli altri risultati dello studio. Inoltre, analizzando la sintomatologia dei soggetti risultati positivi nel corso dello studio, nella Coorte COV002 i partecipanti al gruppo LD/SD sono risultati asintomatici o con sintomi lievi nel 58,9% (N=24) rispetto al 3,8% (N=45) registrato nel gruppo SD/SD.
Le analisi dei sottogruppi, incluse su richiesta dei revisori, pur essendo limitate ai partecipanti di età compresa tra 18 e 55 anni, e ad intervalli tra le 2 dosi (prima e seconda) maggiore di 8 settimane, hanno mostrato risultati simili. Inoltre, i dati preliminari riferiti alle coorti più anziane, mostrano risposte immunitarie simili dopo la vaccinazione con due dosi di ChAdOx1 nCov-19.
I ricercatori sottolineano la necessità di effettuare ulteriori approfondimenti per riuscire a determinare il meccanismo sottostante l’aumentata efficacia del regime LD/SD.
---
Su The Lancet pubblicati i risultati dello studio di fase 2/3 del vaccino sviluppato dalla Oxford University
Sono stati recentemente pubblicati su
The Lancet i
risultati dello studio Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): a single-blind, randomised, controlled, phase 2/3 trial. In questo rapporto vengono descritti i risultati della fase 2 dello studio di fase 2/3 in singolo cieco, randomizzato, controllato (COV002), che ha arruolato adulti sani di età pari o superiore a 18 anni in due centri di ricerca clinica del Regno Unito, stratificati per fascia d’età crescente (18-55 anni, 56-69 anni e con oltre 70 anni). I soggetti risultavano idonei all’arruolamento se non presentavano comorbidità mediche gravi o non controllate oppure un punteggio di fragilità elevato (se di età ≥65 anni). I partecipanti sono stati arruolati in
due gruppi distinti a seconda del dosaggio del vaccino: coorti a basso dosaggio (2.2 × 1010 particelle di virus) e coorti a dose standard (3.5–6.5 × 1010virus). Ciascuna delle coorti a basso dosaggio o a dosaggio standard sono state poi suddivise in dieci gruppi in base alla fascia d’età e alla somministrazione del vaccino sperimentale o di controllo. Nello specifico, per la prima fase di reclutamento dei soggetti che ricevevano un basso dosaggio, sono stati reclutati per primi i partecipanti di età compresa tra 18 e 55 anni, successivamente sono stati reclutati i partecipanti di età compresa tra 56 e 69 anni e un'ulteriore estensione del reclutamento è avvenuta per i soggetti di età superiore a 70 anni solo dopo la revisione indipendente della sicurezza da parte del Data Safety Monitoring Board (DSMB) indipendente. I
gruppi di età compresa tra 18 e 55 anni hanno ricevuto due dosi di vaccino e sono stati assegnati in modo casuale a ricevere il vaccino sperimentale (ChAdOx1 nCoV-19) o il vaccino di controllo (MenACWY). I gruppi di età compresa tra 56 e 69 anni e oltre 70 anni sono stati assegnati in modo casuale a ricevere una o due dosi di vaccino e sono stati quindi assegnati in modo casuale a ricevere il vaccino sperimentale o il vaccino di controllo.
Lo stesso procedimento è stato ripetuto con il reclutamento e la randomizzazione per le coorti a dose standard dopo la revisione da parte del DSMB. Tutti i partecipanti sono stati sottoposti a una visita di screening. Sono stati esclusi dalla partecipazione a tutti i gruppi, i volontari risultati sieropositivi alla SARS-CoV-2 prima dell'arruolamento, ad eccezione di quelli della coorte di 18-55 anni che riceveva una dose standard. Inoltre, tutti i partecipanti inclusi in questa fase 2, ad eccezione di quelli del gruppo a basso dosaggio di 18-55 anni, sono stati sottoposti a ulteriori approfondimenti (esami del sangue per HIV, epatite B e C, emocromo completo e funzionalità epatica e renale). La seconda dose è stata somministrata a 28 giorni di distanza.
I risultati dello studio mostrano che sono stati arruolati 560 partecipanti, di cui 160 di età compresa tra 18 e 55 anni (100 assegnati a ricevere il vaccino ChAdOx1 nCoV-19, 60 assegnati a ricevere MenACWY), 160 di età compresa tra 56 e 69 anni (120 assegnati a ricevere ChAdOx1 nCoV-19 e 40 assegnati a ricevere MenACWY) e 240 di età pari o superiore a 70 anni (200 assegnati a ricevere ChAdOx1 nCoV-19: 40 assegnati a MenACWY). In totale sono stati esclusi 8 partecipanti, di cui 7 non avevano effettuato la seconda dose e 1 aveva ricevuto un dosaggio errato del vaccino. Tre pazienti sono stati invece esclusi dalle analisi relative all’immunogenicità a causa di campioni etichettati in modo errato.
Per quanto concerne le reazioni locali e sistemiche, queste erano più comuni nei partecipanti trattati con ChAdOx1 nCoV-19 rispetto a quelli trattati con il vaccino di controllo ed erano prevalentemente: dolore al sito di iniezione, sensazione di avere la febbre, dolori muscolari, mal di testa, ma erano meno comuni negli anziani (di età ≥56 anni) rispetto ai giovani adulti. A partire dal 26 ottobre 2020, si sono verificati 13 eventi avversi gravi, nessuno dei quali è stato considerato correlato a nessuno dei due vaccini somministrati.
Per quanto riguarda l’immunogenicità, nei partecipanti che avevano ricevuto due dosi di vaccino, entro 14 giorni dalla seconda dose, il 99% dei partecipanti (208 su 209) presentavano risposte anticorpali neutralizzanti. Inoltre, dopo 28 giorni dalla seconda dose sia il picco delle IgG anti-SARS-CoV-2 che gli anticorpi neutralizzanti erano simili nelle tre coorti d’età. Le risposte delle cellule T raggiungevano il picco al 14° giorno dopo una dose singola standard di ChAdOx1 nCoV-19.
In conclusione, secondo gli autori lo studio ha dimostrato che il vaccino ChAdOx1 nCoV-19 sembra essere tollerato meglio negli adulti più anziani rispetto agli adulti più giovani, esso inoltre presenta un’immunogenicità simile nei tre differenti gruppi di età dopo la seconda dose. Sono però necessari ulteriori studi per valutare l’efficacia del vaccino in individui che presentano comorbidità.
---
Lo studio di fase II/III
Investigating a Vaccine Against COVID-19 ha l’obiettivo di determinare l'efficacia, la sicurezza e l'immunogenicità del vaccino candidato contro la malattia da Coronavirus (COVID-19) ChAdOx1 DA -19 in volontari sani del Regno Unito.
E’ previsto l’arruolamento di 12.330 volontari, divisi in 11 gruppi. I gruppi 1, 7 e 9 sono adulti di età compresa tra 56 e 69 anni; i gruppi 2, 8 e 10 sono adulti dai 70 anni in su; il gruppo 3 è composto da bambini di età compresa tra 5 e 12 anni; i gruppi 4, 5, 6 e 11 sono adulti di età compresa tra 18 e 55 anni. Tutti i soggetti saranno sottoposti a follow-up per un 1 anno dopo l'ultima dose di vaccino.
Sponsors and Collaborators: University of Oxford
Ad26.CoV2.S (numero identificativo: NCT04505722)
Il 12 ottobre 2020, a causa dell’insorgenza di una patologia (non prevista) in uno dei partecipanti, l’Azienda farmaceutica
Johnson & Johnson aveva annunciato la momentanea sospensione dello
studio di fase 3 riguardante il
vaccino ricombinante Ad26.COV2.S (ENSEMBLE). I ricercatori, ricordando che studi di grandi dimensioni (lo studio ENSEMBLE prevede la partecipazione di 60mila persone) non sono esenti dal verificarsi di eventi avversi, avevano precisato che
quanto accaduto al partecipante sarebbe stato
sottoposto ad attenta valutazione da parte del Comitato indipendente per il monitoraggio della sicurezza dei dati (DSMB), nonché dal gruppo di sicurezza interno.
--
Lo studio di fase 3
Study of Ad26.COV2.S for the Prevention of SARS-CoV-2-Mediated COVID-19 in Adult Participants (ENSEMBLE) ha lo scopo di dimostrare l'efficacia del vaccino ricombinante Ad26.COV2.S, basato sul vettore adenovirale sierotipo 26 (Ad26) che esprime la proteina spike (S) di SARS-CoV-2, nella prevenzione di COVID-19 mediato da SARS-CoV-2 negli adulti di età pari o superiore a 18 anni. Si tratta di uno studio randomizzato, in doppio cieco, controllato che prevede il coinvolgimento di 60mila partecipanti suddivisi in 178 centri di ricerca. Lo studio prevede due bracci: i partecipanti del
gruppo sperimentale riceveranno l'iniezione intramuscolare (IM) di Ad26.COV2.S con un dosaggio di 1 ^ 10 * 11 particelle virali (vp) come vaccino monodose il giorno 1; i partecipanti del
gruppo di controllo riceveranno un’unica iniezione IM di placebo il giorno 1. EntrambI i gruppi saranno sottoposti ad un periodo di follow-up di 2 anni.
Sponsor and Collaborators: Janssen Vaccines & Prevention B.V.
Ad5-nCoV (numero identificativo: NCT04526990)
Lo studio di fase 3 Phase III Trial of A COVID-19 Vaccine of Adenovirus Vector in Adults 18 Years Old and Above, randomizzato, in doppio cieco, controllato con placebo, ha lo scopo di valutare negli adulti con età ≥ 18 anni l'efficacia, la sicurezza e l'immunogenicità del nuovo vaccino ricombinante (Ad5-nCoV) contro il coronavirus che utilizza un adenovirus (di tipo 5, non replicante) per trasportare il materiale genetico che codifica per la proteina Spike di SARS-CoV-2. Il programma di immunizzazione prevede la somministrazione di una sola dose di vaccino per via intramuscolare nel deltoide. E’ prevista l’arruolamento di 4000 soggetti (2000 nel gruppo sperimentale e 2000 nel gruppo placebo).
Sponsor: CanSino Biologics Inc.
Collaborator: Beijing Institute of Biotechnology
AZD1222 (numero identificativo: NCT04516746)
NovitàAstraZeneca rende noti i dati di efficacia del suo vaccino contro Covid-19: AZD1222 è efficace in media al 70%
Nella lunga corsa alla ricerca di un vaccino efficace contro Covid-19 l’azienda biofarmaceutica AstraZeneca
ha reso noti i primi risultati dell’analisi ad interim della sperimentazione di fase III riguardante il vaccino AZD1222.
La schedula vaccinale di AZD1222 prevede 2 somministrazioni intramuscolari a distanza di 4 settimane. I risultati (ancora non pubblicati) hanno mostrato un'efficacia del 90% quando il vaccino AZD1222 è stato somministrato a dosaggio ridotto (metà dose al tempo 0 e dosaggio pieno a 4 settimane), mentre l’utilizzo del regime standard (due dosi da 5x1010 a distanza di 4 settimane) ha rivelato un’efficacia del 62%. L'analisi combinata di entrambi i regimi di dosaggio (con il coinvolgimento di 11.636 partecipanti) ha prodotto un'efficacia media del 70%.
Tutti i risultati erano statisticamente significativi (p <= 0,0001).
Sul totale dei partecipanti, si sono verificati 131 casi COVID-19 nell'analisi ad interim.
Un comitato di monitoraggio indipendente ha stabilito che l'analisi ha raggiunto il suo endpoint primario mostrando la protezione da COVID-19 che si verifica 14 giorni o più dopo aver ricevuto due dosi del vaccino. Non sono stati confermati eventi gravi di sicurezza riguardanti il vaccino. AZD1222 è stato ben tollerato in entrambi i regimi di dosaggio.
AstraZeneca riprende la sperimentazione clinica del suo vaccino
Riprende la sperimentazione clinica sul vaccino anti-SARS-CoV-2 sviluppato da AstraZeneca e dall'Università di Oxford, dopo essere stata momentaneamente sospesa a causa di un effetto collaterale segnalato in un paziente del Regno Unito.
Dopo un’indagine effettuata da parte di un comitato indipendente, gli esperti e la Medicines Health Regulatory Authority (MHRA), l’ente regolatorio britannico, hanno concluso che è sicuro riprendere la sperimentazione clinica. Oxford e AstraZeneca hanno rifiutato di divulgare qualsiasi informazione medica "per motivi di riservatezza dei partecipanti".
Durante l’interruzione della somministrazione di nuove dosi di vaccino, è continuato regolarmente il monitoraggio dei soggetti a cui il vaccino era già stato somministrato. Complessivamente sono circa 18.000 i soggetti a cui è stato somministrato il vaccino AZD1222.
---
Si tratta di uno studio multicentrico randomizzato, in doppio cieco, controllato con placebo per valutare la sicurezza, l'efficacia e l'immunogenicità del vaccino AZD1222. I partecipanti sono randomizzati in un rapporto 2:1 per ricevere 2 dosi IM di 5 × 10˄10 AZD1222 (n = circa 20.000) o placebo salino (il gruppo di controllo, n = circa 10.000 ) a distanza di 4 settimane l’una dall’altra.
Il vaccino AZD1222 è stato sviluppato congiuntamente dall'Università di Oxford (ChAdOx1 Vector Vaccine) e dalla sua società spin-out, Vaccitech. Nel maggio 2020, l’Azienda AstraZeneca ha ricevuto un ingente finanziamento dalla Biomedical Advanced Research and Development Authority (USA), per lo sviluppo, la produzione e la consegna del vaccino. Lo studio di fase III è parte di questo accordo di finanziamento.
Sponsor: AstraZeneca
Collaborator: Iqvia Pty Ltd
BNT162b2 (numero identificativo: NCT04368728)
Novità L'EMA raccomanda il primo vaccino COVID-19 per l'autorizzazione nell'UE
In data 21 dicembre 2020 l'EMA (European Medicines Agency) ha diramato un
comunicato stampa con cui
raccomanda la
concessione di un'autorizzazione all'immissione in commercio condizionata per il vaccino sviluppato da BioNTech e Pfizer, per prevenire la malattia da coronavirus 2019 (COVID-19) nelle persone a partire dai 16 anni di età. Il parere scientifico dell'EMA apre la strada alla prima autorizzazione all'immissione in commercio di un vaccino COVID-19 nell'UE da parte della Commissione europea, con tutte le garanzie, i controlli e gli obblighi che ciò comporta.
Novità L'FDA rilascia l'autorizzazione all'uso di emergenza per il vaccino Pfizer-BioNTech in soggetti di età pari o superiore a 16 anni
Il vaccino Pfizer-BioNTech è basato sull'antigene della glicoproteina spike (S) di SARS-CoV-2 codificato dall'RNA, formulato in nanoparticelle lipidiche (LNP).
L'uso proposto nell'ambito di un EUA è "per l'immunizzazione attiva per la prevenzione del COVID-19 causato da SARS-CoV-2 in individui di età pari o superiore a 16 anni". Il regime di dosaggio proposto è di 2 dosi, 30 μg ciascuna, somministrate a 21 giorni di distanza. La richiesta EUA include dati di sicurezza ed efficacia provenienti da uno studio in corso sul vaccino BNT162b2 di fase 3, randomizzato, in doppio cieco e controllato con placebo su circa 44.000 partecipanti.
L'endpoint primario di efficacia del vaccino BNT162b2 è stato valutato come l’efficacia nel prevenire l'incidenza di COVID-19 tra i partecipanti senza evidenza di infezione da SARS-CoV-2 prima o durante il regime di vaccinazione a 2 dosi. Il secondo endpoint primario di efficacia è stato valutato come efficacia del vaccino nel prevenire l’incidenza di COVID-19 confermato nei partecipanti con e senza evidenza di precedente infezione da SARS-CoV-2 nei 7 giorni prima della seconda dose. I casi sono stati contati da 7 giorni dopo la seconda dose per entrambi gli endpoint.
Nelle analisi intermedie e finali pianificate, l'efficacia del vaccino dopo 7 giorni la somministrazione della seconda dose è stata del 95% (95% CI 90,3; 97,6) nei partecipanti senza precedente evidenza di infezione da SARS-CoV-2 e > 94% nel gruppo di partecipanti con o senza precedente infezione. I risultati di efficacia erano costantemente robusti (≥93%) nei sottogruppi demografici. L'efficacia contro COVID-19 grave che si verificava dopo la prima dose era dell'88,9%, con un’efficacia del vaccino stimata del 75,0% (1 caso nel gruppo BNT162b2 e 4 casi nel gruppo placebo) contro COVID-19 grave che si verifica almeno 7 giorni dopo la seconda dose. Tra tutti i partecipanti (indipendentemente dall'evidenza di infezione prima o durante il regime di vaccinazione), si sono verificati 50 casi di COVID-19 dopo la prima dose nel gruppo BNT162b2 rispetto a 275 casi nel gruppo placebo, indicando un’efficacia del vaccino stimata dell'82% rispetto a COVID-19 confermato che si verifica dopo la prima dose e con un’efficacia del vaccino del 52,4% tra la prima e la seconda dose. Da un'analisi post-hoc, l'efficacia osservata dopo la prima dose e prima della seconda dose non può supportare evidenze sull'efficacia di una singola dose di vaccino, perché il tempo di osservazione è limitato dal fatto che la maggior parte dei partecipanti ha ricevuto una seconda dose dopo tre settimane e lo studio non prevedeva un braccio monodose per effettuare un confronto adeguato.
Le analisi dei sottogruppi dell'endpoint primario di efficacia hanno mostrato stime di efficacia simili per età, sesso, gruppi razziali ed etnici e partecipanti con comorbidità mediche associate ad alto rischio di COVID-19 grave. In particolare, le analisi dei sottogruppi del secondo endpoint primario di efficacia forniscono ulteriori informazioni sull’efficacia del vaccino per i partecipanti con e senza evidenza di infezione prima della vaccinazione in specifiche popolazioni arruolate, che è l'endpoint considerato rappresentativo della popolazione generale che può ricevere il vaccino, poiché una precedente infezione potrebbe anche non essere nota. Solo il 3% dei partecipanti aveva evidenza di infezione precedente all'arruolamento nello studio e ulteriori analisi hanno mostrato che pochissimi casi di COVID-19 si sono verificati in questi partecipanti nel corso dell'intero studio (9 nel gruppo placebo e 10 nel gruppo BNT162b2, solo 1 dei quali si è verificato a 7 giorni o più dopo il completamento del regime vaccinale). Sebbene limitati, questi dati suggeriscono che individui precedentemente infettati possono essere a rischio di COVID-19 (cioè reinfezione) e potrebbero trarre beneficio dalla vaccinazione. Sono state condotte ulteriori analisi del primo endpoint primario di efficacia per valutare l'efficacia del vaccino in base allo stato di comorbidità. Le stime dell’efficacia del vaccino erano uniformemente alte tra le comorbidità esaminate, sebbene alcune interpretazioni dei risultati siano limitate dal piccolo numero di partecipanti e/o casi.
I dati di sicurezza di circa 38.000 partecipanti ≥16 anni di età randomizzati 1: 1 al vaccino o al placebo con una mediana di 2 mesi di follow-up dopo la seconda dose suggeriscono un profilo di sicurezza favorevole, senza specifici problemi di sicurezza identificati che precluderebbero l'emissione di una EUA . I dati di sicurezza disponibili di tutti i partecipanti arruolati fino al cut-off dei dati del 14 novembre 2020 (N = 43.252, che include l'arruolamento tardivo di ulteriori partecipanti adolescenti e adulti), erano coerenti con il profilo di sicurezza per i circa 38.000 partecipanti con follow-up mediano di 2 mesi e inoltre non avevano sollevato problemi di sicurezza specifici.
Alla data limite, i dati di reattogenicità richiesti nei partecipanti di età compresa tra 16 e 17 anni non sono stati raccolti nel diario elettronico e non sono disponibili. I sintomi coerenti con la reattogenicità sollecitata che sono stati segnalati da questi partecipanti sono stati raccolti e analizzati come eventi avversi non richiesti e sono discussi con la revisione di questi dati.
Le reazioni avverse sollecitate più comuni sono state reazioni nel sito di iniezione (84,1%), affaticamento (62,9%), mal di testa (55,1%), dolore muscolare (38,3%), brividi (31,9%), dolore articolare (23,6%), febbre (14,2 %). L'inizio mediano delle reazioni locali nel gruppo vaccino è stato il giorno della vaccinazione e 2 giorni dopo una delle due dosi ed è durato mediamente tra 1 e 2 giorni. L'insorgenza mediana di eventi avversi sistemici nel gruppo sottoposto a vaccinazione in generale è stata di 1-2 giorni dopo una delle due dosi ed è durato mediamente 1 giorno. La frequenza e la gravità degli eventi avversi sistemici erano maggiori nei gruppi di età più giovane rispetto a quelli più anziani. All'interno di ciascun gruppo di età, la frequenza e la gravità degli eventi avversi sistemici erano maggiori dopo la seconda dose rispetto alla prima dose, ad eccezione di vomito e diarrea, che erano generalmente simili indipendentemente dalla dose. Per entrambi i gruppi di età gli eventi avversi più comuni erano affaticamento, mal di testa e dolore muscolare di nuova insorgenza o peggiorato.
Le reazioni avverse gravi si sono verificate nello 0,0% fino al 4,6% dei partecipanti ed erano più frequenti dopo la seconda dose e generalmente meno frequenti nei partecipanti di età ≥55 anni (≤2,8%) rispetto ai partecipanti più giovani (≤4,6%). La frequenza di eventi avversi gravi è stata bassa (<0,5%), senza differenze significative tra i bracci dello studio. Con l'eccezione di una reattogenicità più frequente, generalmente da lieve a moderata nei partecipanti di età <55 anni, il profilo di sicurezza di BNT162b2 era generalmente simile per gruppi di età, sesso, gruppi etnici e razziali, partecipanti con o senza comorbidità mediche e partecipanti con o senza evidenza di una precedente infezione da SARS-CoV-2 al momento dell'arruolamento.
Sulla base della totalità delle prove scientifiche disponibili, è ragionevole ritenere che il vaccino Pfizer-BioNTech COVID-19 possa essere efficace nella prevenzione del COVID-19 in individui di età pari o superiore a 16 anni, e i benefici noti e potenziali del vaccino Pfizer-BioNTech COVID-19 superano i suoi rischi noti e potenziali per l'uso in individui di età pari o superiore a 16 anni.
Sulla base dei dati disponibili non sembra esserci evidenza di diminuzione della protezione durante un follow-up di circa 2 mesi dopo la seconda dose che viene valutata a questo punto nel tempo.
Il comitato discuterà anche quali studi aggiuntivi dovrebbero essere condotti dal produttore del vaccino dopo l'emissione dell'EUA, per raccogliere ulteriori dati sulla sicurezza e sull'efficacia di questo vaccino.
Novità Pubblicati su NEJM i risultati dello studio di fase III del vaccino Pfizer
In data 10 dicembre è stato pubblicato su The New England Journal of Medicine l'articolo
Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine con i risultati dello studio di fase III del vaccino Pfizer-BioNTech. I risultati erano stati anticipati dalla Pfizer sul proprio sito web, come sotto riportato.
Pfizer pubblica sul proprio sito i risultati preliminari dello studio di fase III, annunciandone la conclusione
La sperimentazione clinica di fase 3 del vaccino BNT162b2 è iniziata il 27 luglio. Il vaccino è stato testato su 43.500 persone in sei Paesi (Stati Uniti, Germania, Turchia, Sud Africa, Brasile e Argentina). Circa il 42% dei partecipanti a livello globale e il 30% dei partecipanti statunitensi appartenevano a gruppi etnici e razziali diversi; il 41% dei partecipanti globali e il 45% dei partecipanti degli Stati Uniti presentavano un'età compresa tra 56 e 85 anni.
Una revisione dei dati di reattogenicità su un sottogruppo randomizzato di almeno 8.000 partecipanti di età pari o superiore a 18 anni nello studio di fase 2/3 ha dimostrato che il vaccino è stato ben tollerato. La maggior parte degli eventi avversi si sono risolti subito dopo la vaccinazione. Non è stato, inoltre, riscontrato nessun serio problema di sicurezza, gli unici eventi avversi di grado 3 con frequenza superiore al 2% sono stati: affaticamento (3,8%) e mal di testa al (2,0%).
Il trial ha dimostrato un’efficacia del 95% contro COVID-19 a partire dal 28° giorno dopo la somministrazione della prima dose di vaccino; sono stati inoltre valutati 170 casi confermati di COVID-19, di cui 162 sono stati osservati nel gruppo placebo, mentre 8 sono stati osservati nel gruppo che aveva ricevuto il vaccino BNT162b2. Quando i partecipanti sono stati stratificati per età, l'efficacia osservata negli adulti di età superiore a 65 anni è risultata superiore al 94%.
Dato che è stato raggiunto il traguardo dei dati di sicurezza richiesto dalla Food and Drug Administration (FDA) statunitense per l'autorizzazione all'uso di emergenza (EUA), le aziende prevedono di presentare entro pochi giorni i dati alla FDA per EUA e di condividerli con altre agenzie di regolamentazione in tutto il mondo.
Le aziende prevedono di produrre a livello globale fino a 50 milioni di dosi di vaccino nel 2020 e fino a 1,3 miliardi di dosi entro la fine del 2021.
---
Risultati preliminari dello studio di fase III sul vaccino anti- SARS-CoV2 sviluppato da Pfizer e BioNTech
La
prima analisi ad interim della fase III dello studio sul vaccino sviluppato da Pfizer e BioNTech mostra che esso può essere efficace in oltre il 90% dei casi nel prevenire la COVID-19 nei partecipanti senza una pregressa infezione.
Un vaccino a mRNA è un vaccino che utilizza dei frammenti di mRNA, i quali contengono unicamente le informazioni per produrre la
glicoproteina Spike (il principale antigene del SARS-CoV2), utilizzato dal virus per agganciarsi ai
recettori ACE2 delle cellule bersaglio.
La produzione degli antigeni stimolerà il sistema immunitario dell'organismo alla proliferazione delle cellule B, che producono anticorpi, e di cellule T, specializzate nel distruggere le cellule infettate dal virus.
---
Novità Il commento su BMJ
E’ stato pubblicato su BMJ un commento di Elisabeth Mahase dal titolo
Covid-19: Vaccine candidate may be more than 90% effective, interim results indicate. In tale commento viene evidenziato che attualmente i risultati dello studio, che sarebbero i primi della fase III di un vaccino anti- SARS-CoV2, sono stati condivisi solo tramite comunicato stampa, ma non sono stati ancora pubblicati o sottoposti a
peer review. Gli esperti hanno accolto con favore la notizia, ma hanno sottolineato la necessità di poter visionare i risultati completi, compresi i dati demografici dei partecipanti, inclusa l’età dei partecipanti allo studio e la gravità degli eventi avversi segnalati. È stata inoltre sottolineata l’importanza di tenere presente che questi sono i primi risultati basati su un numero relativamente ridotto di casi. Inoltre, la stima dell'efficacia si basa su sette giorni di follow-up dei partecipanti dopo la somministrazione della seconda dose; mentre l’analisi dei dati delle prossime settimane e dei prossimi mesi consentirà di delineare un quadro sufficientemente dettagliato sull'efficacia del vaccino a lungo termine. Il vaccino necessita di conservazione a −70 ° C.
Le aziende intenderebbero chiedere l’autorizzazione all'uso di emergenza da parte della US Food e Drug Administration dopo aver raccolto i dati sulla sicurezza del vaccino per i due mesi successivi alla somministrazione della seconda dose. Tali dati sarebbero attesi entro la terza settimana di novembre. Verranno anche pubblicati su rivista peer-reviewed i dati della sperimentazione di fase II.
La sperimentazione clinica proseguirà fino a quando si saranno accumulati un totale di 164 casi confermati di COVID-19 (sviluppati nonostante la vaccinazione), in modo da raccogliere ulteriori dati e valutare altri endpoints della vaccinazione come l’efficacia basata su casi verificatisi 14 giorni dopo la seconda dose.
CoronaVac (numero identificativo: NCT04582344)
Si tratta di uno studio clinico randomizzato, in doppio cieco, controllato con placebo, condotto in adulti di età compresa tra 18 e 59 anni. Lo scopo di questo studio è valutare l'efficacia, la sicurezza e l'immunogenicità del vaccino “CoronaVac”. E’ previsto l’arruolamento di 13.000 partecipanti assegnati a ricevere due dosi di vaccino sperimentale, o placebo, al tempo 0 e 14 giorni dalla prima somministrazione.
Lo studio sarà condotto su due coorti separate:
- la prima sarà formata da operatori sanitari che rappresenteranno il gruppo ad alto rischio (K-1)
- la seconda coorte sarà composta da persone a rischio normale (K-2)
L'analisi preliminare di sicurezza è prevista al completamento del programma vaccinale (2 dosi) in 1.300 volontari (gruppo K-2).
Sponsor: Health Institutes of Turkey
Collaborator: Sinovac Research & Development Co., Ltd.
Gam-COVID-Vac (numero identificativo: NCT04530396)
E’ previsto l’arruolamento di 40.000 soggetti di età superiore ai 18 anni, randomizzati (3:1) in due gruppi:
- un gruppo di riferimento di 10.000 volontari riceveranno il placebo
- un gruppo di studio di 30.000 volontari riceveranno il vaccino sperimentale Gam-COVID-Vac
al tempo 0 e a 21 (±2) giorni dalla prima somministrazione
Ciascun soggetto parteciperà alla sperimentazione per 180 ± 14 giorni dopo la somministrazione della prima dose di vaccino/placebo.
Sponsor: Gamaleya Research Institute of Epidemiology and Microbiology, Health Ministry of the Russian Federation
Novità Vaccino Sputnik V: i dati preliminari mostrano un'efficacia oltre il 95% a 42 giorni dalla prima dose
Gam-COVID-Vac è un vaccino a due vettori basato sull'adenovirus umano che esprime la proteina spike di SARS-CoV-2 per stimolare una risposta immunitaria.
Il vaccino basato sull’adenovirus Ad26 viene utilizzato per la prima dose e il vaccino basato sull’adenovirus Ad5 viene utilizzato il 21° giorno per potenziare la risposta immunitaria.
Attualmente, 40.000 volontari stanno prendendo parte allo studio clinico di fase III (ClinicalTrials.gov: NCT04530396), fra i quali più di 22.000 sono stati vaccinati con la prima dose e 19.000 di questi hanno ricevuto anche la seconda dose. Le sperimentazioni cliniche di fase III sono in corso in Bielorussia, Emirati Arabi Uniti, Venezuela e altri paesi, mentre la fase II-III è in corso in India.
I dati preliminari ottenuti dopo 21 giorni dalla somministrazione della seconda dose (somministrata a 21 giorni dalla prima dose) indicano un'efficacia del vaccino superiore al 95%.
Secondo il protocollo di studio, l’efficacia provvisoria del vaccino SputniK V è calcolata al raggiungimento di 20, 39 e 78 casi di nuove infezioni da coronavirus, registrate sia nel gruppo placebo che nel gruppo che ha ricevuto il vaccino.
Quindi, questa seconda analisi sull'efficacia del vaccino Sputnik V è stata effettuata sulla base di 39 casi confermati, di cui 31 identificati nel gruppo placebo e 8 nel gruppo che ha ricevuto il vaccino. Il rapporto tra il gruppo placebo e il gruppo che ha ricevuto il vaccino è di 1 a 3.
Attualmente non sono stati registrati eventi avversi gravi, mentre alcuni eventi avversi minori includevano dolore nel sito di iniezione e sintomi simil-influenzali, tra cui febbre, debolezza, affaticamento e mal di testa. Il monitoraggio degli aventi avversi è in corso.
La prossima analisi provvisoria dei dati sarà condotta al raggiungimento di 78 casi confermati di coronavirus tra i partecipanti allo studio.
---
Sul sito
sputnikvaccine.com era già stata pubblicata la
prima analisi ad interim riguardante i dati di efficacia. Secondo quanto riportato sul sito, sulla base della prima analisi ad interim ottenuta 21 giorni dopo la prima iniezione, l'
efficacia del vaccino Sputnik V è stata del
92% (calcolo basato sui 20 casi confermati di COVID-19 suddivisi tra individui vaccinati e coloro che hanno ricevuto il placebo). È in corso il monitoraggio dei partecipanti ed al momento non ci sono stati eventi avversi inattesi durante gli studi clinici.
---
Dopo il completamento delle sperimentazioni cliniche di fase III del vaccino Sputnik V, il Centro Gamaleya fornirà l'accesso al rapporto completo della sperimentazione clinica.
mRNA 1273 (numero identificativo: NCT04470427)
Novità L'FDA rilascia l'autorizzazione all'uso di emergenza per il vaccino Moderna in soggetti con età pari o superiore a 18 anni
---
L’azienda Moderna ha lavorato a stretto contatto con National Institutes of Health (NIH), Biomedical Advanced Research and Development (BARDA) e Operation Warp Speed (OWS) allo sviluppo del vaccino mRNA-1273.
mRNA-1273 è un nuovo vaccino a base di acido ribonucleico messaggero (mRNA), incapsulato in una nanoparticella lipidica (LNP). In particolare, il vaccino mRNA-1273 è attualmente in fase di valutazione:
- nello studio 301, di fase 3, randomizzato, in cieco per l'osservatore, controllato con placebo, finalizzato a valutare la sicurezza, l’efficacia e l’immunogenicità di mRNA-1273 alla dose di 100 µg
- nello studio 201 di fase 2a, randomizzato, cieco per l'osservatore e controllato con placebo finalizzato alla valutazione della sicurezza e immunogenicità di 50 µg e 100 µg di mRNA-1273
- nello studio 101 di fase 1, in aperto, a dosaggio variabile di 25 µg, 50 µg, 100 µg o 250 µg finalizzato a valutare la sicurezza e l’immunogenicità dell'mRNA 1273.
I risultati dell’immunogenicità dello studio 101 hanno mostrato che il vaccino alla dose di 100 μg somministrata in due iniezioni a 28 giorni di distanza, inducono la formazione di anticorpi neutralizzanti in tutti i partecipanti a partire dalla prima settimana dopo la seconda iniezione.
La risposta immunitaria è risultata simile tra i gruppi di età persistendo per almeno 3 mesi dopo la seconda iniezione. Pertanto, la dose da 100 µg di mRNA-1273 era stata selezionata come dose ottimale negli studi successivi.
I risultati dell'immunogenicità dello studio 201 hanno confermato l'induzione del legame degli anticorpi e degli anticorpi neutralizzanti contro la proteina spike SARS-CoV-2 sia alla dose di 50 μg che a quella di 100 μg.
Dato che lo studio 301 presenta più del 96% dei dati di sicurezza prodotti fino ad oggi con mRNA-1273, il
documento informativo mRNA Moderna Covid-19 Vaccine si concentra sui dati disponibili e provenienti da questo studio. Nello studio 301 sono stati arruolati 30.418 partecipanti di età pari o superiore a 18 anni a maggior rischio di contrarre COVID-19, assegnati in modo casuale in un rapporto 1: 1 a ricevere 2 iniezioni di 100 μg di mRNA-1273 o placebo, ciascuno somministrato in due iniezioni a 28 giorni di distanza. La randomizzazione è stata stratificata in base all'età e alla presenza o assenza di fattori di rischio per malattia grave COVID-19 (in base alla raccomandazione CDC di marzo 2020) nei partecipanti <65 anni di età. I partecipanti erano considerati a rischio di grave malattia da COVID-19 se avevano almeno 1 dei seguenti fattori di rischio allo screening:
• Malattia polmonare cronica (es. enfisema e bronchite cronica), idiopatica (fibrosi polmonare e fibrosi cistica) o asma da moderata a grave;
• Malattia cardiaca significativa (es. insufficienza cardiaca, malattia coronarica, malattie cardiache congenite, cardiomiopatie e ipertensione polmonare);
• Grave obesità (indice di massa corporea ≥ 40 kg / m2)
• Diabete (tipo 1, tipo 2 o gestazionale)
• Malattia del fegato
• Infezione da virus dell'immunodeficienza umana (HIV) controllata
L'endpoint primario di efficacia era rappresentato dalla valutazione di efficacia del vaccino mRNA-1273 per prevenire il primo evento di COVID-19 e l'analisi includeva i casi che iniziavano 14 giorni dopo la seconda iniezione, valutati da un comitato indipendente di valutazione in cieco rispetto all'assegnazione del gruppo di vaccini. Gli endpoint secondari di efficacia, anch'essi giudicati da un comitato indipendente di valutazione, includevano la valutazione dell’efficacia del vaccino mRNA-1273 a prevenire quanto segue:
• Grave COVID-19
• COVID-19 indipendentemente dalla sintomatologia o dalla gravità, definita come prima insorgenza di infezione da SARS-CoV-2 o COVID-19 a partire da 14 giorni dopo la seconda dose di vaccino
• COVID-19 basato su una definizione secondaria (meno restrittiva) a partire da 14 giorni dopo la seconda iniezione di vaccino. La definizione secondaria (meno restrittiva) di COVID-19 includeva uno dei seguenti sintomi: febbre (temperatura ≥ 38 ° C) o brividi, tosse, mancanza di respiro o difficoltà a respirare, affaticamento, dolori muscolari, mal di testa, nuova insorgenza di perdita del gusto o dell'olfatto, mal di gola, congestione nasale o rinorrea, nausea o vomito o diarrea e un tampone nasale o campione di saliva (o campione respiratorio, se ricoverato in ospedale) positivo per SARS-CoV-2 mediante RT-PCR.
• Morte dovuta a COVID-19
• COVID-19 a partire da 14 giorni dopo la prima iniezione di vaccino (inclusi i casi che sono avvenuti dopo la seconda iniezione).
Un endpoint secondario aggiuntivo era basato sul set FAS (Full Analysis Set): la valutazione dell’efficacia di mRNA-1273 nel prevenire COVID-19 indipendentemente dalla precedente infezione da SARS-CoV-2 ,14 giorni dopo la seconda dose di vaccino.
L'analisi primaria ha mostrato che l’efficacia del vaccino mRNA-1273 nel prevenire i sintomi COVID-19 nei partecipanti al basale sieronegativi per COVID-19, era del 94,1%. In base alla valutazione effettuata da comitato di valutazione indipendente, a partire da 14 giorni dopo la seconda dose di vaccino, si sono verificati 196 casi di COVID-19.
Questi risultati hanno confermato l'analisi ad interim di efficacia, basata sui 95 casi, con 5 casi che si sono verificati nel gruppo mRNA-1273 e 90 casi che si sono verificati nel gruppo placebo. Nell'analisi ad interim, la stima puntuale della valutazione dell’efficacia era del 94,5%, coerente con l’efficacia rilevata nell’analisi primaria. I risultati della valutazione di efficacia del vaccino a mRNA-1273 dell’endpoint secondario erano simili all’endpoint primario, con stime puntuali di valutazione dell’efficacia compresa tra il 93,6% e il 100% in base agli hazard ratio. Al momento dell’analisi primaria, i casi riportati di COVID-19 grave sono stati 30 tutti verificatisi nel gruppo placebo. Di conseguenza la stima puntuale dell'efficacia contro la malattia grave è risultata del 100%. Sfortunatamente, uno di questi casi ha provocato un decesso dovuto a COVID-19 in un soggetto del gruppo placebo.
Per quanto concerne gli eventi avversi locali o sistemici, sono stati osservati nella maggior parte dei partecipanti sottoposti alla vaccinazione con mRNA-1273 e generalmente sono aumentate dopo la seconda iniezione. Le reazioni locali e sistemiche dopo l’iniezione erano più alte nel gruppo vaccino rispetto al gruppo placebo. La maggior parte degli eventi avversi nel gruppo mRNA-1273 erano di grado da 1 a 2 (secondo scale di valutazione che vanno da 0 a 4) e generalmente si risolvevano entro 3 giorni o meno. I tassi di incidenza degli eventi avversi durante i 28 giorni successivi all'iniezione sono stati generalmente simili nei partecipanti che hanno ricevuto mRNA-1273 e in quelli che hanno ricevuto placebo.
Nello specifico, per quanto riguarda le reazioni locali con insorgenza entro 7 giorni dopo l’iniezione (es. giorno di somministrazione e 6 giorni dopo, ma nel caso in cui un evento avverso persisteva più di 7 giorni al partecipante veniva chiesto di continuare a registrare le reazioni fino a risoluzione dell’evento) includevano: dolore, eritema, gonfiore e linfadenopatia, nessuna reazione locale di grado 4 è stata sollecitata, e solo il dolore di grado 3 è stato segnalato con una frequenza inferiore al 2% dopo ciascuna iniezione.
Per quanto riguarda le reazioni sistemiche, queste includevano: febbre, mal di testa, affaticamento, mialgia, artralgia, brividi e nausea / vomito, erano prevalenti nel gruppo mRNA-1273 rispetto al gruppo placebo. Nel gruppo mRNA-1273, l'incidenza e la gravità delle reazioni sistemiche sembravano aumentare dopo la seconda iniezione. La maggior parte delle reazioni sistemiche erano di grado da 1 a 2 di gravità. La maggior parte delle reazioni sistemiche sollecitate nei partecipanti che hanno ricevuto mRNA-1273 si sono verificate entro i primi 1 o 2 giorni dall'iniezione e generalmente sono continuate mediamente da 1 a 2 giorni.
Gli eventi avversi che persistevano oltre i 7 giorni erano simili tra mRNA-1273 e placebo, senza alcuna differenza notevole tra la prima e la seconda iniezione.
In conclusione, l'efficacia di mRNA-1273 è risultata simile tra i principali gruppi demografici e sottogruppi ad aumentato rischio di COVID-19 grave e morte.
Vaccino Moderna, l'azienda fa sapere che è «efficace al 100% contro Covid grave»
La biotech statunitense ha annunciato il 30 novembre 2020 che l’analisi primaria dello studio di Fase 3 su mRNA-1273, condotto su 196 casi, conferma l’elevata efficacia osservata alla prima analisi ad interim. L’analisi dei dati indica un’efficacia del vaccino pari al 94,1% ed un'efficacia risultata pari al 100% nell’endopoint dello studio che ha valutato i casi gravi di COVID-19: se ne sono verificati 30 e tutti nel gruppo di controllo. Sulla base di questi risultati, Moderna procede a chiedere oggi stesso un’autorizzazione all’uso di emergenza (EUA) dalla Food and Drug Administration e l’approvazione condizionale dell’Agenzia europea per i medicinali. Consulta anche la nota adnkronos.
L’analisi ad interim, analizzando i dati provenienti da circa 30.000 partecipanti arruolati in 100 centri di ricerca clinica negli Stati Uniti, ha riferito che il potenziale vaccino è risultato sicuro e ben tollerato con un tasso di efficacia pari al 94,5%.
La schedula vaccinale, definita nello studio, prevede la somministrazione intramuscolare di due dosi vaccinali a distanza di 29 giorni (T0 e T29 giorni). L’azienda, inoltre, ha annunciato che il vaccino mRNA-1273 presenta una stabilità a temperature standard comprese fra -2° e -8°C per 30 giorni facilitando, così, la procedura di conservazione, distribuzione e somministrazione (leggi la
news anche sul sito del National Institute of Health - NIH)
L’
Agenzia europea del farmaco (EMA) ha avviato la
procedura di revisione progressiva dei risultati, che continuerà fino a quando non saranno disponibili prove sufficienti a supportare i criteri previsti per la formale autorizzazione all'immissione in commercio. L'EMA valuterà la conformità del vaccino ai consueti standard di efficacia, sicurezza e qualità. Sebbene la tempistica complessiva della revisione non possa ancora essere prevista, il processo dovrebbe essere più breve di una valutazione regolare per il tempo guadagnato visto che la valutazione dei risultati avviene con frequenza continua.
---
I partecipanti ricevono 1 iniezione intramuscolare (IM) di 100 microgrammi (ug) di mRNA-1273 il giorno 1 e il giorno 29 e sono confrontati con il gruppo che riceve il placebo (iniezione di cloruro di sodio allo 0,9%, soluzione salina normale).
Sponsor: ModernaTX, Inc.
Collaborators: Biomedical Advanced Research and Development Authority, National Institute of Allergy and Infectious Diseases (NIAID)
Nanoparticle Vaccine With Matrix-M1TM Adjuvant (EudraCT Number: 2020-004123-16)
E’ previsto l’arruolamento di 9.000 partecipanti (randomizzati in due gruppi) assegnati a ricevere due dosi di vaccino sperimentale, o placebo, al tempo 0 e a 21 giorni dalla prima somministrazione.
Sponsor: Novavax
PROFISCOV (numero identificativo: NCT04456595)
E’ attualmente in fase III Clinical Trial of Efficacy and Safety of Sinovac's Adsorbed COVID-19 (Inactivated) Vaccine in Healthcare Professionals (PROFISCOV) la sperimentazione clinica per valutare l'efficacia e la sicurezza del vaccino anti COVID-19 (inattivato, adsorbito) negli operatori sanitari. Lo studio sarà in doppio cieco controllato con placebo con partecipanti assegnati in modo casuale 1: 1 ai bracci placebo e vaccino. Il programma di immunizzazione prevede iniezioni intramuscolari di due dosi con un intervallo di 14 giorni. Per valutare la sicurezza e immunogenicità, i partecipanti saranno divisi in due gruppi di età, adulti (18-59 anni) e anziani (60 anni e oltre). Il database sulla sicurezza ha come obiettivo quello di rilevare reazioni avverse con frequenza di 1: 1000 o superiore negli adulti e di 1: 500 negli anziani.
Tutti i partecipanti saranno seguiti fino a 12 mesi. L'analisi preliminare dell'efficacia sarà avviata raggiungendo il numero target di 150 casi.
Sponsor: Butantan Institute
Collaborator: Sinovac Life Sciences Co., Ltd.
Vero cell (numero identificativo: NCT04560881)
Lo studio prevede il coinvolgimento di 3.000 partecipanti sani di età compresa tra 18 e 85 anni, assegnati a ricevere in modo casuale (randomizzato) due dosi di vaccino sperimentale Vero Cell, o placebo, al tempo 0 e dopo 21 giorni dalla prima somministrazione.
Sponsor: Laboratorio Elea Phoenix S.A.
Collaborators: Beijing Institute of Biological Products Co., Ltd.
A cura di:
Caterina Silvestri, Agenzia regionale di sanità della Toscana
Cristina Stasi, Centro Interdipartimentale di Epatologia CRIA-MASVE, Dipartimento di Medicina Sperimentale e Clinica, AOU Careggi

Nessun commento:
Posta un commento