

La teoria è che il virus, che è stato sviluppato da esperti in malattie infettive per funzionare come una bio-arma, è nato nel laboratorio con sede a Wuhan del Dr. Peng Zhou , il principale ricercatore cinese del sistema immunitario dei pipistrelli, in particolare nel modo in cui i loro sistemi immunitari si adattano alla presenza di virus come il coronavirus e altri virus distruttivi. In qualche modo, il virus è fuggito dal laboratorio e il mercato del pesce di Hunan, dove si presume che il virus abbia avuto origine, è solo uno stratagemma.
Ora, un rispettato epidemiologo che di recente ha preso piede per aver affermato in una minaccia di Twitter che il virus sembrava essere molto più contagioso di quanto si pensasse inizialmente sta sottolineando irregolarità nel genoma del virus che suggerisce che potrebbe essere stato geneticamente modificato ai fini di un'arma, e non solo qualsiasi arma, ma la più mortale di tutte.
In " Strana somiglianza di inserti unici nella proteina del picco 2019-nCoV con HIV-1 gp120 e Gag ", i ricercatori indiani sono sconcertati da segmenti dell'RNA del virus che non hanno alcuna relazione con altri coronavirus come la SARS, e invece sembrano essere più vicini a HIV. Il virus risponde anche al trattamento con farmaci per l'HIV.

Per coloro che hanno poco tempo a disposizione, ecco i principali risultati del documento, che si concentra prima sulla natura unica del 2019-nCoV, quindi osservano quattro sequenze di aminoacidi nel Coronavirus di Wuhan che sono omologhe alle sequenze di aminoacidi nell'HIV1:
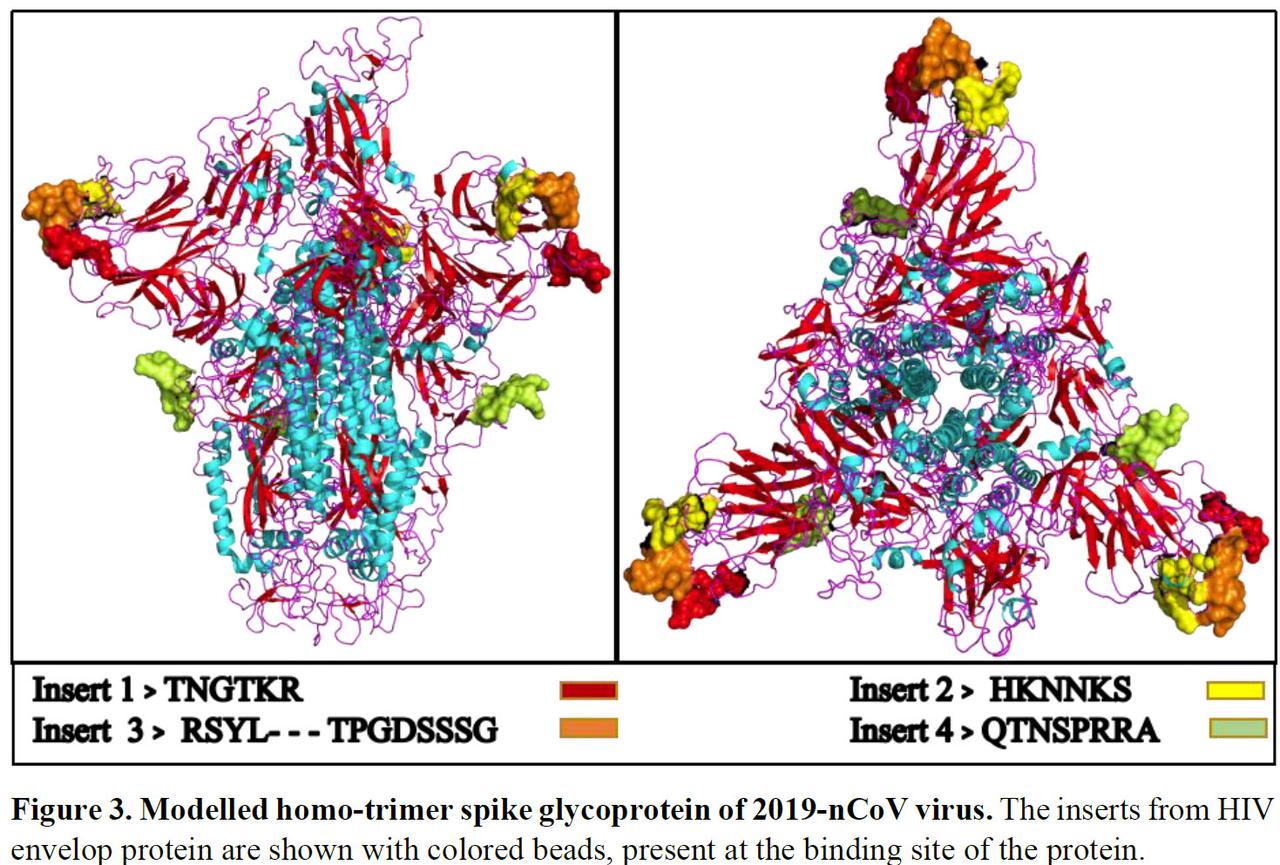
Il nostro albero filogenetico di coronavirus a lunghezza intera suggerisce che 2019-nCoV è strettamente correlato al SARS CoV [Fig1].Inoltre, altri studi recenti hanno collegato il 2019-nCoV al SARS CoV. Abbiamo quindi confrontato le sequenze di glicoproteine di picco del 2019-nCoV con quelle del SARS CoV (numero di adesione NCBI: AY390556.1). Su un attento esame dell'allineamento della sequenza abbiamo scoperto che la glicoproteina con spike 2019-nCoV contiene 4 inserzioni [Fig.2]. Per indagare ulteriormente se questi inserti sono presenti in qualsiasi altro virus corona, abbiamo eseguito un allineamento di sequenze multiple delle sequenze di amminoacidi glicoproteina spike di tutti i coronavirus disponibili (n = 55) [fare riferimento a Tabella S.1] in NCBI refseq (ncbi.nlm .nih.gov) include una sequenza di 2019-nCoV [Fig.S1]. Abbiamo scoperto che questi 4 inserimenti [inserti 1, 2, 3 e 4] sono unici per 2019-nCoV e non sono presenti in altri coronavirus analizzati. Un altro gruppo cinese ha documentato tre inserzioni confrontando un minor numero di sequenze di glicoproteine con spike di coronavirus. Un altro gruppo cinese ha documentato tre inserzioni confrontando un minor numero di sequenze di glicoproteine di spike di coronavirus (Zhou et al., 2020).Abbiamo quindi tradotto il genoma allineato e abbiamo scoperto che questi inserti sono presenti in tutti i virus Wuhan 2019-nCoV ad eccezione del virus 2019-nCoV di Bat come host [Fig.S4]. Incuriositi dai 4 inserti altamente conservati esclusivi di 2019-nCoV, volevamo capire la loro origine. A tale scopo, abbiamo utilizzato l'allineamento locale 2019-nCoV con ciascun inserto come query contro tutti i genomi dei virus e considerato hit con una copertura della sequenza del 100%. Sorprendentemente, ciascuno dei quattro inserti si è allineato con brevi segmenti delle proteine Human Immunficiency Virus-1 (HIV-1) . Le posizioni degli aminoacidi degli inserti nel 2019-nCoV e i corrispondenti residui in HIV-1 gp120 e HIV-1 Gag sono mostrati nella Tabella 1.I primi 3 inserti (inserire 1,2 e 3) allineati a brevi segmenti di residui di aminoacidi nell'HIV-1 gp120. L'inserto 4 allineato al bavaglio dell'HIV-1. L'inserto 1 (6 residui di amminoacidi) e l'inserto 2 (6 residui di amminoacidi) nella glicoproteina spike del 2019-nCoV sono identici al 100% ai residui mappati sull'HIV-1 gp120. L'inserto 3 (12 residui di amminoacidi) nel 2019- nCoV è mappato all'HIV-1 gp120 con lacune [vedi tabella 1]. L'inserto 4 (8 residui di aminoacidi) è mappato al bavaglio dell'HIV-1 con lacune.

Perché gli autori pensano che il virus possa essere creato dall'uomo? Perché quando si osservano gli inserimenti di cui sopra che non sono presenti in nessuna delle famiglie di coronavirus più vicine, "è abbastanza improbabile che un virus abbia acquisito inserimenti così unici naturalmente in un breve periodo di tempo". Invece, possono essere trovati nell'identificazione delle cellule e nelle proteine leganti la membrana situate nel genoma dell'HIV.
Poiché la proteina S del 2019-nCoV condivide la più vicina origine con SARS GZ02, la codifica in sequenza per le proteine di picco di questi due virus è stata confrontata utilizzando il software MultiAlin. Abbiamo trovato quattro nuovi inserimenti nella proteina del 2019-nCoV- “GTNGTKR” (IS1), “HKNNKS” (IS2), “GDSSSG” (IS3) e “QTNSPRRA” (IS4) (Figura 2). Con nostra sorpresa, questi inserimenti di sequenza non erano solo assenti nella proteina S della SARS, ma non sono stati osservati in nessun altro membro della famiglia dei Coronaviridae (figura supplementare). Ciò è sorprendente poiché è abbastanza improbabile che un virus abbia acquisito inserimenti così unici naturalmente in un breve periodo di tempo.È stato osservato che gli inserimenti sono presenti in tutte le sequenze genomiche del virus 2019-nCoV disponibili dai recenti isolati clinici. Per conoscere l'origine di questi inserimenti nel 2019-nCoV è stato fatto un allineamento locale con BLASTp usando questi inserimenti come query con tutto il genoma del virus. Inaspettatamente, tutte le inserzioni sono state allineate con il virus dell'immunodeficienza umana-1 (HIV-1). Ulteriori analisi hanno rivelato che sequenze allineate di HIV-1 con 2019-nCoV sono state derivate dalla glicoproteina di superficie gp120 (posizioni di sequenza degli aminoacidi: 404-409, 462-467, 136-150) e dalla proteina Gag (366-384 aminoacido) ( Tabella 1).La proteina gag dell'HIV è coinvolta nel legame con la membrana ospite, nel confezionamento del virus e nella formazione di particelle simili al virus. Gp120 svolge un ruolo cruciale nel riconoscere la cellula ospite legandosi al recettore primario CD4. Questo legame induce riarrangiamenti strutturali in GP120, creando un sito di legame ad alta affinità per un co-recettore delle chemochine come CXCR4 e / o CCR5.
E alcuni elementi visivi, che portano gli autori della carta a concludere che "questo cambiamento strutturale potrebbe anche aver aumentato la gamma di cellule ospiti che 2019-nCoV può infettare" :
La modellazione 3D della struttura proteica ha mostrato che questi inserimenti sono presenti nel sito di legame di 2019-nCoV. A causa della presenza di motivi gp120 nella glicoproteina spike 2019-nCoV nel suo dominio di legame, proponiamo che questi inserimenti di motivi potrebbero aver fornito una maggiore affinità con i recettori delle cellule ospiti. Inoltre, questo cambiamento strutturale potrebbe anche aver aumentato l'intervallo di cellule ospiti che 2019-nCoV può infettare . Per quanto ne sappiamo, la funzione di questi motivi non è ancora chiara nell'HIV e deve essere esplorata. Lo scambio di materiale genetico tra i virus è ben noto e tale scambio critico evidenzia il rischio e la necessità di studiare le relazioni tra famiglie di virus apparentemente non correlate.
Un buon riassunto delle scoperte è stato fornito dal dott. Feigl-Ding, che ha iniziato la sua discussione esplicativa sottolineando che la velocità di trasmissione al di fuori della Cina ha superato quella all'interno della Cina.


2) Whoa- the rate of increase ***outside of China*** is steeper than inside of China or Wuhan! Figure 1A. From: @TheLancet “Nowcasting and forecasting the potential domestic and international spread of 2019-nCoV
”)

2) Whoa- the rate of increase ***outside of China*** is steeper than inside of China or Wuhan! Figure 1A. From: @TheLancet “Nowcasting and forecasting the potential domestic and international spread of 2019-nCoV
”)
 media. (75k estimate from above Lancet article)
media. (75k estimate from above Lancet article)
Ma la "pistola fumante" in questo caso sono parti del codice genetico del virus che i ricercatori indiani, guidati da Prashant Pradhan presso l'Indian Institute of Technology, hanno scoperto che potrebbero essere stati "incorporati" dall'HIV, che appartiene a una famiglia di virus completamente diversa .


17. ...WHOA- the authors said the finding was “Unexpectedly” related to genes from HIV virus. Notably there were 4 gene insertions (see figure in above post #16). And so, which HIV gene proteins were found in the new #coronarvirus? Gag protein and Gp120- key HIV proteins...

19. Again, these are new express published findings and not peer reviewed yet. Let’s not draw conclusions yet. But evidence suggest that 2 different HIV genesare present in the #coronarvirus S gene region (that didn’t map to any other coronavirus, according to other studies).


 What a bold paper... I don’t know what to say
What a bold paper... I don’t know what to say ")

La battuta finale:
9. BOTTOMLINE: 1) Seafood market not the source. 2) This RNA #coronavirus mutates really fast. 3)


Un altro dottore intervenne con quella che pensava fosse una solida spiegazione delle irregolarità del virus ...


... Fino a quando non ha realizzato qualcosa di inquietante.

"Spaventoso" ... ma rilassati, è solo un'altra ridicola "cospirazione". Fonte: qui
Quanto sarà grave l'epidemia di coronavirus?
Ecco 6 fattori chiave
Mentre l'epidemia di coronavirus continua a diffondersi in tutta la Cina, una raffica di prime ricerche sta disegnando un quadro più chiaro di come si comporta l'agente patogeno e dei fattori chiave che determineranno se può essere contenuto.
-
Sembra moderatamente contagioso, simile alla SARS.
-
È ancora difficile da sapere. Ma il tasso di mortalità è probabilmente inferiore al 3 percento, molto inferiore alla SARS.
-
Forse tra 2 e 14 giorni, permettendo alla malattia di non essere rilevata.
-
Il virus si diffuse rapidamente perché è iniziato in un hub di trasporto.
-
L'OMS ha elogiato gli sforzi della Cina, ma i critici temono che le misure di blocco potrebbero non essere sufficienti.
-
Un vaccino è ancora lontano un anno - almeno.
Mentre il virus è una grave preoccupazione per la salute pubblica, il rischio per la maggior parte delle persone al di fuori della Cina rimane molto basso e l'influenza stagionale è una minaccia più immediata. Per evitare qualsiasi malattia virale, gli esperti consigliano di lavarsi le mani frequentemente ed evitare il proprio ufficio o scuola quando si è malati. La maggior parte delle persone sane non ha bisogno di maschere e l'accumulo di queste può contribuire alla carenza di operatori sanitari che ne hanno bisogno, dicono gli esperti.
Quanto è contagioso il virus?
Sembra moderatamente contagioso, simile alla SARS.
La portata di un focolaio dipende dalla rapidità e dalla facilità con cui un virus viene trasmesso da persona a persona. Mentre la ricerca è appena iniziata, gli scienziati hanno stimato che ogni persona con il coronavirus di Wuhan potrebbe infettare da qualche parte tra 1,5 e 3,5 persone senza efficaci misure di contenimento.
Ciò renderebbe il nuovo virus più o meno contagioso come la SARS, un altro coronavirus che è circolato in Cina nel 2003 ed è stato contenuto dopo aver ammalato 8.098 persone e ucciso 774. Virus respiratori come questi possono viaggiare nell'aria, avvolti in minuscole goccioline che vengono prodotte quando una persona malata respira, parla, tossisce o starnutisce.
These droplets fall to the ground within a few feet. That makes the virus harder to get than pathogens like measles, chickenpox and tuberculosis, which can travel a hundred feet through the air. But it is easier to catch than H.I.V. or hepatitis, which spread only through direct contact with the bodily fluids of an infected person.
How far viruses travel

Coronaviruses like the Wuhan virus can travel only about six feet from the infected person. It’s unknown how long they live on surfaces.
Some other viruses, like measles, can travel up
to 100 feet and stay alive on surfaces for hours.
If each person infected with the Wuhan coronavirus infects two to three others, that may be enough to sustain and accelerate an outbreak, if nothing is done to reduce it.
Ecco come funziona. Nell'animazione di seguito, un gruppo di cinque persone infette potrebbe diffondere il virus a circa 368 persone in soli cinque cicli di infezione.
Se 5 persone con coronavirus di Wuhan hanno contagiato 2.6 altre ...
... potrebbero esserci5 people sick dopo1 cycle .
Confrontalo con un virus meno contagioso, come l'influenza stagionale. Le persone con influenza tendono a infettare 1,3 altri individui, in media. La differenza può sembrare piccola, ma il risultato è un notevole contrasto: solo circa 45 persone potrebbero essere infettate nello stesso scenario.
Se 5 persone con influenza stagionale contagiassero 1.3 altre ...
... potrebbero esserci5 people sick dopo1 cycle .
Ma i numeri di trasmissione di qualsiasi malattia non sono fissati nella pietra. Possono essere ridotti mediante efficaci misure di sanità pubblica, come l'isolamento dei malati e il monitoraggio delle persone con cui hanno avuto contatti. Quando le autorità sanitarie globali hanno sistematicamente rintracciato e isolato le persone infette da SARS nel 2003, sono state in grado di portare a 0,4 il numero medio di persone malate infette, abbastanza da fermare l'epidemia.
Le autorità sanitarie di tutto il mondo stanno facendo enormi sforzi nel tentativo di ripeterlo.
Finora il numero di casi al di fuori della Cina è stato limitato. Ma nei giorni scorsi sono comparsi casi in diversi paesi, tra cui gli Stati Uniti, con persone che non hanno visitato la Cina. E il numero di casi in Cina è aumentato, superando di gran lunga il tasso di nuovi casi di SARS nel 2003:

10.000 casi segnalati
Wuhan
coronavirus
8,000
6,000
SARS
4,000
2,000
I numeri sono aumentati dopo la SARS
casi dalla terraferma
Sono stati segnalati Cina.
0
20o giorno
40
60
80
100
120
140
Il primo giorno in cui l'OMS ha ricevuto segnalazioni di focolai
Note: il conteggio dei casi ufficiali dell'Organizzazione mondiale della sanità per la SARS è stato ritardato all'inizio dell'epidemia. Alcuni casi sono stati sospettati ma non confermati; La SARS è una diagnosi di esclusione, quindi i casi precedentemente segnalati potrebbero essere stati scartati dopo ulteriori indagini. Dati sul coronavirus di Wuhan alle 23:30 ET del 30 gennaio.
Quanto è mortale il virus?
È ancora difficile da sapere. Ma il tasso di mortalità è
probabilmente inferiore al 3 percento, molto inferiore alla SARS.
Questo è uno dei fattori più importanti per quanto sarà dannoso l'epidemia e uno dei meno compresi.
È difficile valutare la mortalità di un nuovo virus. I casi peggiori vengono di solito rilevati per primi, il che può distorcere la nostra comprensione della probabilità che i pazienti muoiano. Circa un terzo dei primi 41 pazienti segnalati a Wuhan ha dovuto essere trattato in terapia intensiva, molti con sintomi di febbre, tosse grave, respiro corto e polmonite. Ma le persone con casi lievi potrebbero non visitare mai un medico. Quindi potrebbero esserci più casi di quelli che conosciamo e il tasso di mortalità potrebbe essere inferiore a quanto inizialmente pensato.
Allo stesso tempo, i decessi per virus potrebbero essere sottostimati. Le città cinesi al centro dell'epidemia sono carenti di kit di test e letti ospedalieri e molti malati non sono stati in grado di consultare un medico.
"C'è ancora molta incertezza su come sia questo virus e cosa stia facendo", ha dichiarato Allison McGeer, specialista in malattie infettive al Mount Sinai Hospital di Toronto, che era in prima linea nella risposta canadese alla SARS.
Le prime indicazioni suggeriscono che il tasso di mortalità per questo virus è considerevolmente inferiore rispetto a un altro coronavirus, MERS, che uccide circa una persona su tre che viene infettata, e SARS, che ne uccide circa uno su 10. Tutte le malattie sembrano aggrapparsi alle proteine la superficie delle cellule polmonari, ma MERS e SARS sembrano essere più distruttivi per il tessuto polmonare. Al 31 gennaio, meno di uno su 40 delle persone con infezioni confermate era morto. Molti di quelli che morirono erano uomini più anziani con problemi di salute di base.
Ecco come il nuovo coronavirus si confronta con altre malattie infettive:

Tasso di mortalità
(scala del registro)
100%
Influenza aviaria
Ebola
WALKING
50
Vaiolo
Di Più
mortale
20
Influenza spagnola
Polio
10
SARS
5
Coronavirus di Wuhan
La maggior parte delle stime indicano che
tasso di mortalità inferiore al 3%,
e il numero di
trasmissioni tra
1.5 e 3.5.
2
Si diffonde più velocemente
1
Influenza suina
Morbillo
0.1
di stagione
influenza
Comune
freddo
Varicella
0
1
5
10
15
Numero medio di persone infette da ciascun malato
Nota: vengono mostrati i tassi medi di mortalità per caso e i numeri di trasmissione. Le stime dei tassi di mortalità per caso possono variare e i numeri per il coronavirus di Wuhan sono stime preliminari.
Il grafico sopra usa una scala verticale logaritmica : i dati nella parte superiore sono compressi in uno spazio più piccolo per rendere più facile vedere la variazione tra malattie meno mortali. Le malattie nella parte superiore del grafico sono molto più mortali di quelle nel mezzo.
Gli agenti patogeni possono ancora essere molto pericolosi anche se il loro tasso di mortalità è basso, ha detto la dott.ssa McGeer. Ad esempio, anche se l'influenza ha un tasso di mortalità inferiore a uno su 1.000, circa 200.000 persone finiscono in ospedale con il virus ogni anno negli Stati Uniti e circa 35.000 persone muoiono.
Quanto tempo ci vuole per mostrare i sintomi?
Forse tra 2 e 14 giorni, permettendo alla malattia di non essere rilevata.
Il tempo necessario affinché compaiano i sintomi dopo che una persona è stata infettata può essere vitale per la prevenzione e il controllo. Conosciuto come periodo di incubazione, questa volta può consentire ai funzionari sanitari di mettere in quarantena o osservare le persone che potrebbero essere state esposte al virus. Ma se il periodo di incubazione è troppo lungo o troppo breve, queste misure potrebbero essere difficili da attuare.
Alcune malattie, come l'influenza, hanno un breve periodo di incubazione di due o tre giorni. Le persone possono rilasciare particelle di virus infettivo prima di manifestare sintomi influenzali, rendendo quasi impossibile identificare e isolare le persone che hanno il virus. La SARS, tuttavia, ha avuto un periodo di incubazione di circa cinque giorni. Inoltre, ci sono voluti quattro o cinque giorni dopo l'inizio dei sintomi prima che i malati potessero trasmettere il virus. Ciò ha dato ai funzionari il tempo di fermare il virus e contenere efficacemente l'epidemia, ha affermato il dott. McGeer.
I funzionari dei Centri statunitensi per il controllo e la prevenzione delle malattie stimano che il coronavirus di Wuhan abbia un periodo di incubazione da 2 a 14 giorni. Ma non è ancora chiaro se una persona possa diffondere il virus prima che si manifestino i sintomi o se la gravità della malattia influenzi la facilità con cui un paziente può diffondere il virus.
"Questo mi preoccupa perché significa che l'infezione potrebbe eludere il rilevamento", ha dichiarato il dott. Mark Denison, esperto di malattie infettive alla Vanderbilt University di Nashville.
Quanto hanno viaggiato le persone infette?
Il virus si diffuse rapidamente perché è iniziato in un hub di trasporto.
Wuhan è un posto difficile per contenere un focolaio. Ha 11 milioni di persone, più di New York City. In un giorno medio, 3.500 passeggeri effettuano voli diretti da Wuhan verso città di altri paesi. Queste città sono state tra le prime a segnalare casi di virus al di fuori della Cina.
Passeggeri che volano da Wuhan verso altri paesi
Da ottobre a novembre 2019

Russia
Regno Unito
Italy
Francia
Unito
stati
8,000
Giappone
23,000
viaggiatori
CINA
tacchino
Wuhan
Corea del Sud
Taiwan
Emirati Arabi Uniti
Filippine
Malaysia
Tailandia
55,000
Indonesia
5,000
25,000
Australia
50,000
viaggiatori
Nota: la mappa mostra il volume dei passeggeri da ottobre a novembre 2019, i dati più recenti disponibili.
Wuhan è anche un importante snodo di trasporti all'interno della Cina, collegato a Pechino, Shanghai e altre città importanti da ferrovie ad alta velocità e compagnie aeree nazionali. In ottobre e novembre dello scorso anno, quasi due milioni di persone hanno volato da Wuhan verso altre località della Cina.

Pechino
136,000
CINA
Wuhan
Shanghai
119,000
Passeggeri che volano da Wuhan
in altre città della Cina
Da ottobre a novembre 2019
5,000
25,000
Hong Kong
18,000
Kunming
95.000 passeggeri
50,000
viaggiatori
Nota: la mappa mostra il volume dei passeggeri da ottobre a novembre 2019, i dati più recenti disponibili. Le destinazioni con meno di 1.000 passeggeri non vengono visualizzate.
La Cina non era altrettanto ben collegata nel 2003 durante l'epidemia di SARS. Un gran numero di lavoratori migranti ora viaggia a livello nazionale e internazionale - verso l'Africa, altre parti dell'Asia e dell'America Latina, dove la Cina sta facendo un'enorme spinta infrastrutturale con la sua iniziativa Belt and Road. Questo viaggio crea un alto rischio di epidemie in paesi con sistemi sanitari che non sono attrezzati per gestirli, come lo Zimbabwe, che sta affrontando un peggioramento della fame e una crisi economica.
Soprattutto, la Cina ha circa quattro volte più passeggeri del treno e del trasporto aereo rispetto allo scoppio della SARS:

4 miliardi di viaggiatori
Il traffico passeggeri è quadruplicato, aprendo più rotte per l'infezione.
3
Quando scoppiò la SARS, c'erano circa 1 miliardo di viaggiatori.
2
Treno
viaggiatori
1
Passeggeri aerei
1990
’00
’10
2019
Nota: i dati sui viaggi aerei comprendono i passeggeri solo sulle compagnie aeree cinesi.
La Cina ha fatto il passo senza precedenti di imporre restrizioni di viaggio a decine di milioni di persone che vivono a Wuhan e nelle città vicine. Ma gli esperti hanno avvertito che il blocco potrebbe essere arrivato troppo tardi e l'accesso limitato a cibo e medicine. Il sindaco di Wuhan ha riconosciuto che cinque milioni di persone avevano lasciato la città prima dell'inizio delle restrizioni, in vista del capodanno lunare.
“Non puoi salire su un germe. Si diffonderà una nuova infezione ”, ha affermato Lawrence O. Gostin, professore di legge presso la Georgetown University e direttore del Centro di collaborazione dell'Organizzazione mondiale della sanità sulla legislazione nazionale e globale sulla salute. “Uscirà; lo fa sempre. "
Quanto sarà efficace la risposta?
L'OMS ha elogiato gli sforzi della Cina, ma i critici
temono che le misure di blocco potrebbero non essere sufficienti.
Oltre a chiudere i trasporti, i funzionari hanno chiuso un mercato a Wuhan che vendeva pollame vivo, frutti di mare e animali selvatici, che si pensava fosse l'origine del coronavirus, e in seguito ha sospeso il commercio di animali selvatici a livello nazionale. Le scuole sono state chiuse, la Grande Muraglia di Pechino è vietata e pacchetti turistici dalla Cina sono stati fermati. I funzionari dell'Organizzazione mondiale della sanità hanno elogiato la risposta aggressiva della Cina al virus.
Ma le misure hanno avuto anche effetti indesiderati. I residenti a Wuhan che non stanno bene devono camminare o andare in bicicletta per chilometri per raggiungere gli ospedali. Lì, molti si lamentano del fatto che vengono respinti a causa della carenza di letti, personale e forniture ospedaliere che sono stati aggravati dal blocco.
Fino a poco tempo fa, i ricercatori all'estero erano anche preoccupati del fatto che la Cina non ammettesse esperti che potevano aiutare a rintracciare il virus e prevenirne la diffusione.
Giovedì, l'OMS ha dichiarato l'epidemia di coronavirus di Wuhan un'emergenza sanitaria globale , riconoscendo che la malattia rappresenta un rischio oltre la Cina.
Funzionari sanitari negli Stati Uniti e in altri paesi hanno iniziato a controllare i passeggeri in arrivo negli aeroporti e a isolare coloro che sembrano essere malati. Diversi paesi, tra cui Kazakistan, Russia e Vietnam, hanno temporaneamente limitato i viaggi e i visti da e verso la Cina. Ma i critici temono che queste misure non saranno sufficienti.
Quanto tempo ci vorrà per sviluppare un vaccino?
Un vaccino è ancora lontano un anno - almeno.
Un vaccino contro il coronavirus potrebbe prevenire le infezioni e fermare la diffusione della malattia. Ma i vaccini richiedono tempo.
Dopo l'epidemia di SARS nel 2003, i ricercatori hanno impiegato circa 20 mesi per preparare un vaccino per i test sull'uomo. (Il vaccino non è mai stato necessario, perché alla fine la malattia è stata contenuta.) A partire dall'epidemia di Zika nel 2015, i ricercatori avevano ridotto la cronologia di sviluppo del vaccino a sei mesi.
Ora, sperano che il lavoro delle epidemie passate contribuirà a ridurre ulteriormente i tempi. I ricercatori hanno già studiato il genoma del nuovo coronavirus e hanno scoperto le proteine che sono cruciali per l'infezione. Gli scienziati del National Institutes of Health, in Australia e almeno tre società stanno lavorando su candidati vaccinali.
"Se non incontriamo ostacoli imprevisti, saremo in grado di avviare una sperimentazione di Fase 1 entro i prossimi tre mesi", ha dichiarato il dott. Anthony Fauci, direttore del National Institute of Allergy and Malattie infettive.
Nessun commento:
Posta un commento